
Abstract
Background:
Previous in vitro experiments have demonstrated that prostaglandin F2-alpha (PF2α) reduced proliferation and adipogenesis in a murine cell line and human orbital fibroblasts derived from subjects with inactive Graves' orbitopathy (GO). The objective of this study was to determine if the PGF2α analogue bimatoprost is effective at reducing proptosis in this population.
Methods:
A randomized controlled double-masked crossover trial was conducted in a single tertiary care academic medical center. Patients with long-standing, inactive GO but persistent proptosis (>20 mm in at least one eye) were recruited. Allowing for a 15% dropout rate, 31 patients (26 females) were randomized in order to identify a treatment effect of 2.0 mm (p = 0.05; power 0.88). Following informed consent, participants were randomized to receive bimatoprost or placebo for three months, after which they underwent a two-month washout before switching to the opposite treatment. The primary outcome was the change in exophthalmometry readings over the two three-month treatment periods.
Results:
The mean exophthalmometer at baseline was 23.6 mm (range 20.0–30.5 mm), and the mean age of the patients was 55 years (range 28–74 years). The median duration of GO was 7.6 years (interquartile range 3.6–12.3 years). The majority were still suffering from diplopia (61.3%) with bilateral involvement (61.3%). Using multi-level modeling adjusted for baseline, period, and carry-over, bimatoprost resulted in a −0.17 mm (reduction) exophthalmometry change ([confidence interval −0.67 to +0.32]; p = 0.490). There was a mean change in intraocular pressure of −2.7 mmHg ([confidence interval −4.0 to −1.4]; p = 0.0070). One patient showed periorbital fat atrophy on treatment, which resolved on stopping treatment. Independent analysis of proptosis by photographic images (all subjects) and subgroup analysis on monocular disease (n = 12) did not show any apparent benefit.
Conclusions:
In inactive GO, bimatoprost treatment over a three-month period does not result in an improvement in proptosis.
Introduction
Graves' orbitopathy (GO) is the commonest extrathyroidal manifestation of Graves' hyperthyroidism. Proptosis may persist after inflammation has subsided in the late “burnt out” phase of GO, and the persistent disfigured appearance of the eyes is a source of significant psychological distress and impaired quality of life for sufferers (1). There are no specific medical treatments that target orbital volume reduction in late-stage disease. A British nationwide survey of patients with GO revealed low satisfaction levels with existing therapies (2).
The main pathological features of GO include expansion of orbital tissue fat and muscle, mononuclear cell infiltration of orbital connective tissue and extraocular muscle, and tissue remodeling, a process that can culminate in fibrosis and diminished eye motility (3). A key mechanism underlying GO is an increase in adipogenesis and muscle-associated secretion of glycosaminoglycans in the orbit, resulting in an increase in orbital volume and exophthalmos (protrusion of the eye) (4,5). The opposite effect, enophthalmos (recession of the eye into the orbit), has been described in patients with glaucoma treated with daily bimatoprost (prostaglandin F2-alpha [PGF2α]), a prostaglandin analogue used topically in the management of intraocular hypertension (glaucoma). Cases of enophthalmos developing in patients treated with bimatoprost and other PGF2α analogues have been reported worldwide, albeit in small numbers (6 –10). This side effect is more noticeable if only one eye is exposed to treatment, as the treated eye is easily comparable to the unexposed eye. However, since most patients receive treatment to both eyes, it is possible that the incidence of enophthalmos in bimatoprost-treated patients has been underestimated.
A possible mechanism by which PGF2α agonists might produce enophthalmos is through reduction of orbital fat volume (6). A PGF2α receptor agonist has been shown to be a potent inhibitor of adipose tissue differentiation in newborn rat precursor cells (11). This raises the possibility that PGF2α exerts direct effects on adipose tissue precursors. This finding has been confirmed in in vitro studies using 3T3-L1 cell lines and human primary orbital fibroblast cultures (12). This is further supported by Eftekhari et al. who reported that retrobulbar bimatoprost injections in rats showed histologic evidence of orbital fat atrophy (13). Thus, PGF2α agonists may be effective in reducing orbital fat expansion, ameliorating proptosis, and thus improving quality of life in patients with active and/or inactive disease.
Rehabilitative surgery is the mainstay of treatment for the late disease phase. However, surgery is not always successful in reducing proptosis and carries the associated risks of anesthesia and local complications (14). Recently, teprotumumab, a human monoclonal antibody inhibitor of insulin-like growth factor 1 receptor (IGF-1R) has been shown to reduce proptosis in patients with active GO (15), while radiotherapy is of questionable benefit in conjunction with steroids (16). However, there remains a major unmet need for medical therapies to reduce residual proptosis in the late phase (inactive) of GO, a disease stage in which disfigurement and impairment of ocular function persist after resolution of the initial inflammatory process and which affects 5–10 times as many people as the early active phase (17). In an earlier in vitro study, the majority of samples studies were from patients with inactive GO, and PGF2α was noted to reduce proliferation and adipogenesis in orbital fibroblasts from both GO and non-GO tissue (12). Even in “burnt out” disease, orbital fibroblasts from GO have higher proliferation and adipogenesis potential than cells from normal orbits (12). Therefore, a randomized double-masked crossover clinical study was designed to evaluate the impact of bimatoprost at reducing proptosis in patients with GO.
Methods
The trial was conducted according to the protocol and in compliance with the principles of the Declaration of Helsinki (1996), the principles of Good Clinical Practice, and in accordance with Medicines for Human Use (Clinical Trials) Regulations 2004, as amended in 2006, the Research Governance Framework for Health and Social Care, the Data Protection Act 1998, and other regulatory requirements as appropriate. The trial was approved by a local NHS Research Ethics Committee (registration number: 14/WA/0081) and the Medicines and Healthcare Products Regulatory Agency (registration number: 21323/0043/001-0001), and is registered with
This was a single-center randomized controlled double-masked crossover trial of bimatoprost in GO. Allocation of subjects was by remote computerized Web-based randomization and minimization over two identified factors (degree of proptosis and uni/bilateral eye involvement) to ensure a balance between the two trial arms.
Patients were recruited from the multidisciplinary GO clinic at the University Hospital of Wales (Cardiff, United Kingdom). All patients had had a previous diagnosis of GO defined by the presence of one or more of the following features: soft-tissue changes in the eye, proptosis, extraocular muscle dysfunction, corneal abnormalities, and optic-nerve involvement. The inclusion criteria were: stable GO with no reported change in proptosis for at least six months, inactive disease with a clinical activity score <3, proptosis (subjective unilateral proptosis confirmed by asymmetry in exophthalmometry of >2 mm or >20 mm on exophthalmometry measurement in one eye), euthyroid (free triiodothyronine [fT3] and free thyroxine [fT4] in the reference range), and, if female, using a reliable form of contraception during the trial. The exclusion criteria were: younger than 18 years of age; dysthyroid optic neuropathy; pregnancy/lactation; receiving therapy for glaucoma; systemic steroid use; patients with risk factors for cystoid macular edema, iritis, or uveitis; and allergies to bimatoprost or preservative. Patients were assessed at a screening visit at least two weeks prior to a first trial visit to ensure that they had inactive disease. Patients were allocated either bimatoprost or placebo for three months, followed by a two-month washout period before crossing over to the opposite treatment. Bimatoprost 0.03% (Lumigan®; Allergan) or placebo (Blumont Healthcare) was administered at a dose of one drop in the affected eye/eyes once daily between 6:00pm and midnight, starting from the day of allocation. To enhance masking, the placebo contained artificial tears with a similar preservative (benzalkonium chloride), which will replicate any mild stinging sensation experienced with bimatoprost. Patients were allowed to use preservative-free eye drops for symptomatic dry eyes if needed during the trial, which had to be applied at least 30 minutes before/after application of the trial drops. No other eye drops were allowed during the trial period.
The primary outcome was the change in proptosis with bimatoprost using the mean improvement of the two eyes where both were treated. A change of >2.0 mm in proptosis is considered to be clinically relevant (18,19). Assuming a standard deviation of 2.5 mm in proptosis measurements in patients with GO, as previously reported (19,20), it was calculated that 26 participants would be needed to be able to identify a treatment effect of 2.0 mm as statistically significant (p = 0.05; power 0.88). Allowing for a 15% dropout rate/incomplete data sets, 31 participants were recruited.
At each visit, patients underwent ophthalmological assessment, including assessment of proptosis (using an Oculus exophthalmometer [Supplementary Fig. S1 and Table S1]), intraocular pressure (IOP) in primary position and up gaze, logMAR visual acuity, clinical activity score, palpebral aperture, Gorman's diplopia score, corneal integrity, quality-of-life questionnaires (GO quality of life questionnaire [GO-QOL] and EQ-5D-5L), and a health economic assessment using a modified client service receipt inventory for GO (see Supplementary Data—BIMA study protocol). Color photographs of the eye in the lateral and anterior views were taken according to a standard operating procedure (see Supplementary Data—standard operating procedure). Photograph exophthalmometry measurements were made following 200% magnification from standard view from either the lateral canthus or the nasal bridge to the corneal apex by a masked assessor. Any adverse events were recorded in the patient's diary. Thyroid function tests (thyrotropin, fT3, and fT4) were performed at the beginning, middle, and end of trial visits to ensure patients remained euthyroid. Secondary outcomes were a change in GO-QOL, a change in IOP in primary and chin-forward positions, side-effect profiles of bimatoprost, and health economic evaluation. The ophthalmology assessment was carried out by either one of two assessing ophthalmologists. An initial exophthalmometer alignment phase was conducted whereby the assessors were calibrated by multiple exophthalmometer readings on the same non-trial subjects in the clinic, and adjustments were made to ensure their readings were comparable. Subjects were not necessarily assessed by the same assessor at each time point. In order to ensure maintenance of masking, during each trial visit, the assessors did not have access to baseline values or any prior measurements and clinical notes.
The mean change in proptosis measurement in the placebo phase and bimatoprost phase was compared using a paired t-test. This was carried out using the mean improvement of the two eyes where both were treated or the change in one eye where only one was treated. The multi-level model in STATA v12.1 (StataCorp) using demographic and clinical variables (including baseline, the order of treatment, and carry-over effects) was also used to adjust for unexplained variance and in order to obtain better estimates of effect size with tighter confidence intervals. The results are expressed as an effect in millimeters from the treatment arm controlling for the placebo effect with confidence intervals (CI) and p-values. Secondary and other outcomes were summarized with descriptive statistics.
Three patients were deemed to be protocol non-compliant with inclusion criteria who had fT4 levels above the reference range with normal fT3 during the screening period. This was due to a misinterpretation of the inclusion criteria requiring both a normal fT4 and fT3 (rather than either fT4 or fT3). These three patients were clinically euthyroid during randomization. A sensitivity analysis was done after the exclusion of these three subjects to determine any effect on the study conclusions.
Results
Recruitment and retention
Seventy-two patients were invited initially of whom 33 agreed to trial enrolment. One patient was ineligible on screening, and one patient chose not to take part due to fear that bimatoprost might change her iris color. Thirty-one patients were subsequently randomized and underwent the first phase of the trial successfully. Unfortunately, one patient from the bimatoprost starting group died at the end of the first washout period due to a pulmonary embolism, which was not considered to be related to the investigational product. Therefore, 30 patients were entered into the second phase of the trial. One patient from the placebo starting group did not return for visit 4 (end of second-phase assessment) due to the withdrawal of consent. Twenty-nine patients entered the second washout phase and completed the trial (Fig. 1).
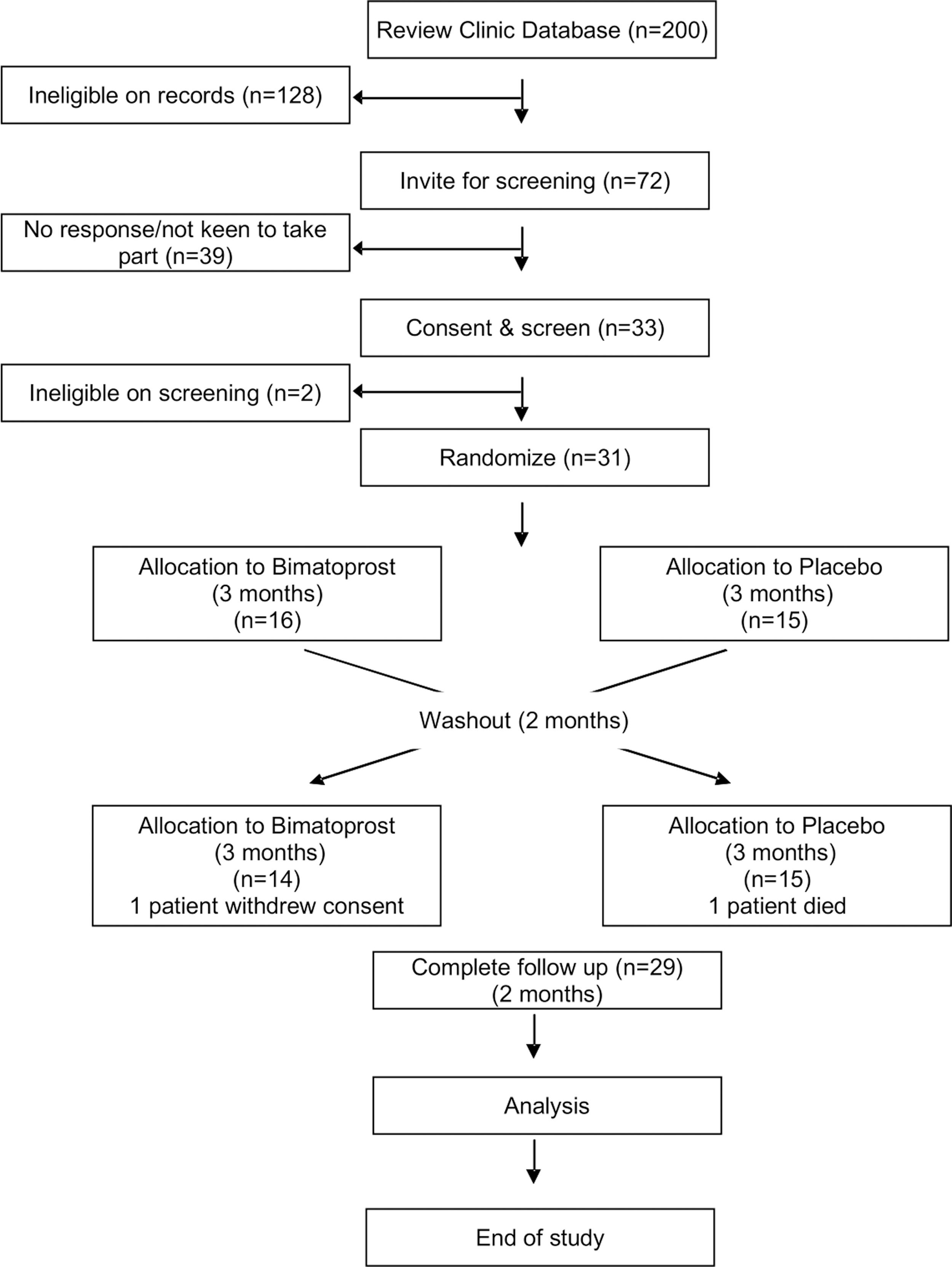
Study consort diagram. Trial participants were randomized to receive Bimatoprost or placebo (n = number of participants at each stage). One patient from Bimatoprost starting group died during washout period from pulmonary embolism and one patient from placebo starting group did not complete final two trial visits.
Demographic and baseline characteristics
Of 31 patients, there was a female preponderance with a 5:1 ratio and a mean age of 55.2 years (range 28–74 years). The median duration of GO was 7.6 years (interquartile range 3.6–12.4 years). The majority were smokers at diagnosis (74.2%), but this reduced to 38.7% after the diagnosis. Of the 31 patients, 19 (61%) were still suffering from diplopia, and 19 (61%) had bilateral involvement. There was a good balance between the two treatment allocations, with some differences in smoking history but not at trial entry, and more patients with constant diplopia in the bimatoprost starting group (Table 1). Thyroid function tests were unchanged throughout the study period.
Baseline Demographics of the Study Population
Data presented as means (standard deviation or range) unless stated otherwise or % (patient number/total). The diplopia severity was assessed by Gorman score and GO severity according to European Group on Graves' Orbitopathy criteria.
BMI, body mass index; IQR, interquartile range; GO, Graves' orbitopathy; fT4, free thyroxine (9.0–19.1 pmol/L); TSH, thyrotropin (0.30–4.4 mIU/L).
Inter-operator comparison
Fifteen non-trial patients were assessed by the two assessors by exophthalmometry after a period of calibration between assessors involving five patients. Compared to assessor 1, the regression coefficient of assessor 2 was 0.93 mm [CI 0.83–1.03 mm]. There was a positive Pearson correlation (r = 0.9652; p < 0.0001) between the two assessors (Supplementary Figs. S2 and S3).
Primary outcome analysis
The mean baseline exophthalmometer readings of treated eyes in the bimatoprost starting group was 24.1 mm (standard deviation = 2.9 mm) and 23.1 (standard deviation = 1.9 mm) in the placebo starting group (Table 1). The mean change across all affected eyes in the Bimatoprost phase was +0.17 mm [CI −0.35 to +0.69] versus +0.26 mm [CI −0.51 to +1.03] in the placebo phase. This was not statistically different (p = 0.845; Fig. 2). A sensitivity analysis was done after exclusion of the three protocol non-compliant subjects. There was no difference between the two groups (p = 0.727). Using the pkcross function in STATA, no period (p = 0.38) or carry-over (p = 0.46) effects were observed.
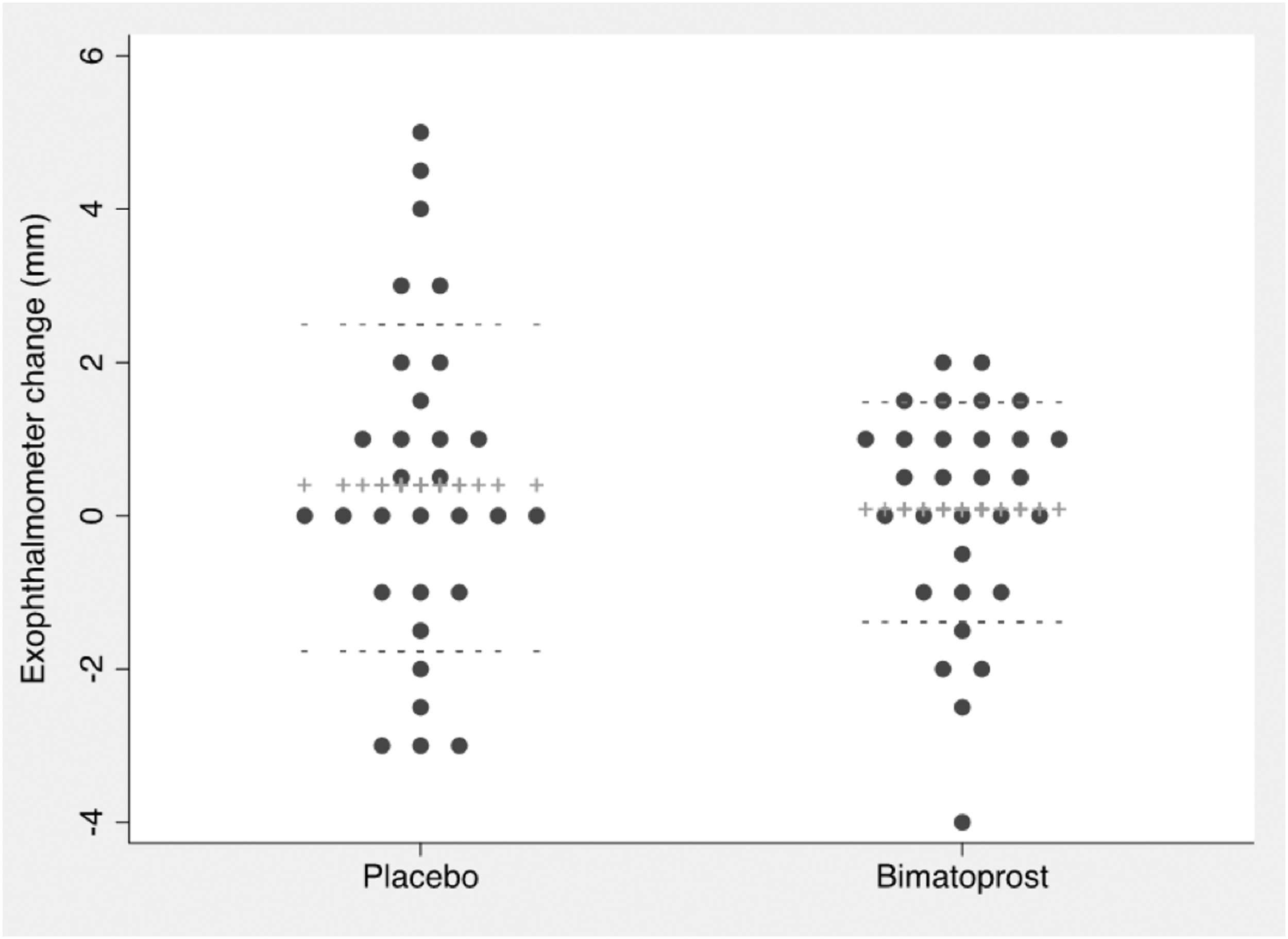
Dot plot of the mean change in proptosis measurement in the placebo phase and bimatoprost phase expressed in millimeters. The mean change in measurement was calculated by subtracting the baseline measurement from that following treatment. Therefore, negative value indicates an improvement in the treatment. ++, the mean; −−, standard deviation. Paired t-test p-value = 0.8455.
Multi-level modeling
Data were also analyzed using a multi-level model in STATA, which also enabled one data point to be used for those patients who were unwilling or unable to proceed to the second phase of the protocol, thus using all available data as efficiently as possible. In this process, each patient's measured eye outcome was nested within each individual patient.
Crude analysis adjusted for baseline (model 1) did not show any treatment effect on the exophthalmometer readings, with a coefficient of −0.27 mm ([CI −1.43 to +0.89]; p = 0.648). Adding multi-level modeling correcting for baseline and phase of treatment (model 2) resulted in a treatment coefficient of −0.17 mm [CI −0.67 to +0.32], which again was not statistically significant (p = 0.490). Carry-over adjustment was omitted because of collinearity with the phase of treatment. Adding the assessors to the model did not improve the model, with a treatment effect of −0.16 mm ([CI −0.65 to 0.33]; p = 0.531) and an assessor coefficient of −0.34 mm ([CI −0.96 to 0.27]; p = 0.274). Removing three patients with protocol deviation resulted in a model 2 treatment coefficient of −0.06 mm ([CI −0.56 to +0.45]; p = 0.827) and a model 3 treatment coefficient of −0.04 mm ([CI −0.55 to +0.46]; p = 0.861). Using response to a 10% drop in IOP as a surrogate marker for compliance showed no statistically significant treatment effect on proptosis as measured on the exophthalmometer (Table 2).
Beta Coefficient of Bimatoprost Effect on Exophthalmometer Readings Using Multi-Level Modeling with Each Treated Patient's Eye
Minus protocol deviation indicated three patients removed from the analysis due to the stated reason. Minus IOP non-responder indicated removal of eyes from analysis with at least a 10% reduction in IOP (surrogate marker to compliance). Model 1 adjusted for baseline; Model 2 adjusted for baseline, phase, and carry-over; Model 3 adjusted for baseline phase, carry-over, and assessors.
CI, confidence interval; IOP, intraocular pressure.
Exophthalmometer change in patients with unilateral proptosis
There were 12 patients with unilateral proptosis. In these patients, only one eye with proptosis was treated, while the other eye served as a control. Analysis of the exophthalmometer reading revealed predicted baseline exophthalmometer differences, with a higher exophthalmometer mean in the treated eye of 22.17 mm [CI 21.16–23.17] versus 20.33 mm [CI 19.14–21.52] in the untreated eye (p = 0.0032). Treatment with bimatoprost did not result in a statistically significant reduction in exophthalmometer results, with a mean change of +0.08 mm [CI −0.66 to +0.82] in the treated eye compared to 0.67 mm [CI −0.58 to +1.92] in the untreated eye (p = 0.1516).
Exophthalmometry and photographic assessment correlations
Proptosis measurements were also made by photographic assessment of the patient photos taken during the trial. The measurements were taken from either the lateral canthus or the nasal bridge to the corneal apex by a masked assessor (Fig. 3). All data from five visits were used for this analysis. Results of the Spearman correlation indicated that there was a significant positive association between exophthalmometer and lateral canthus measurements (Spearman's ρ = 0.609; p < 0.0001). There was a significant negative correlation between exophthalmometer and nasal bridge measurements (Spearman's ρ = −0.396; p < 0.0001; see Figs. 4 and 5). The latter finding was expected, as the measurement was taken from the nasal bridge to the corneal apex (i.e., the more proptosis, the lesser the distance between the corneal apex to the nasal bridge).
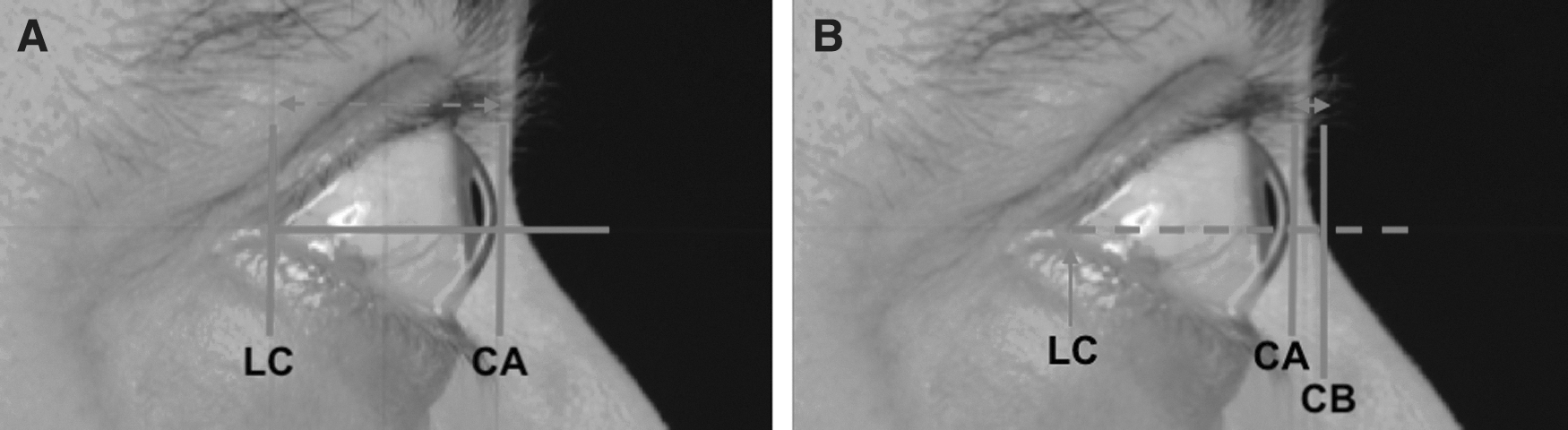
Photographic exophthalmometry measurement following 200% magnification from standard view from the lateral canthus (LC) or the nasal bridge (NB) to the corneal apex (CA) using the LC as a referral point. A horizontal line was drawn from the LC, and a vertical line was drawn perpendicular to the horizontal line where it meets the CA. The distance (left-right dashed arrow) from the LC to the crossed section point is then measured on the screen to obtain the LC to CA measurement (
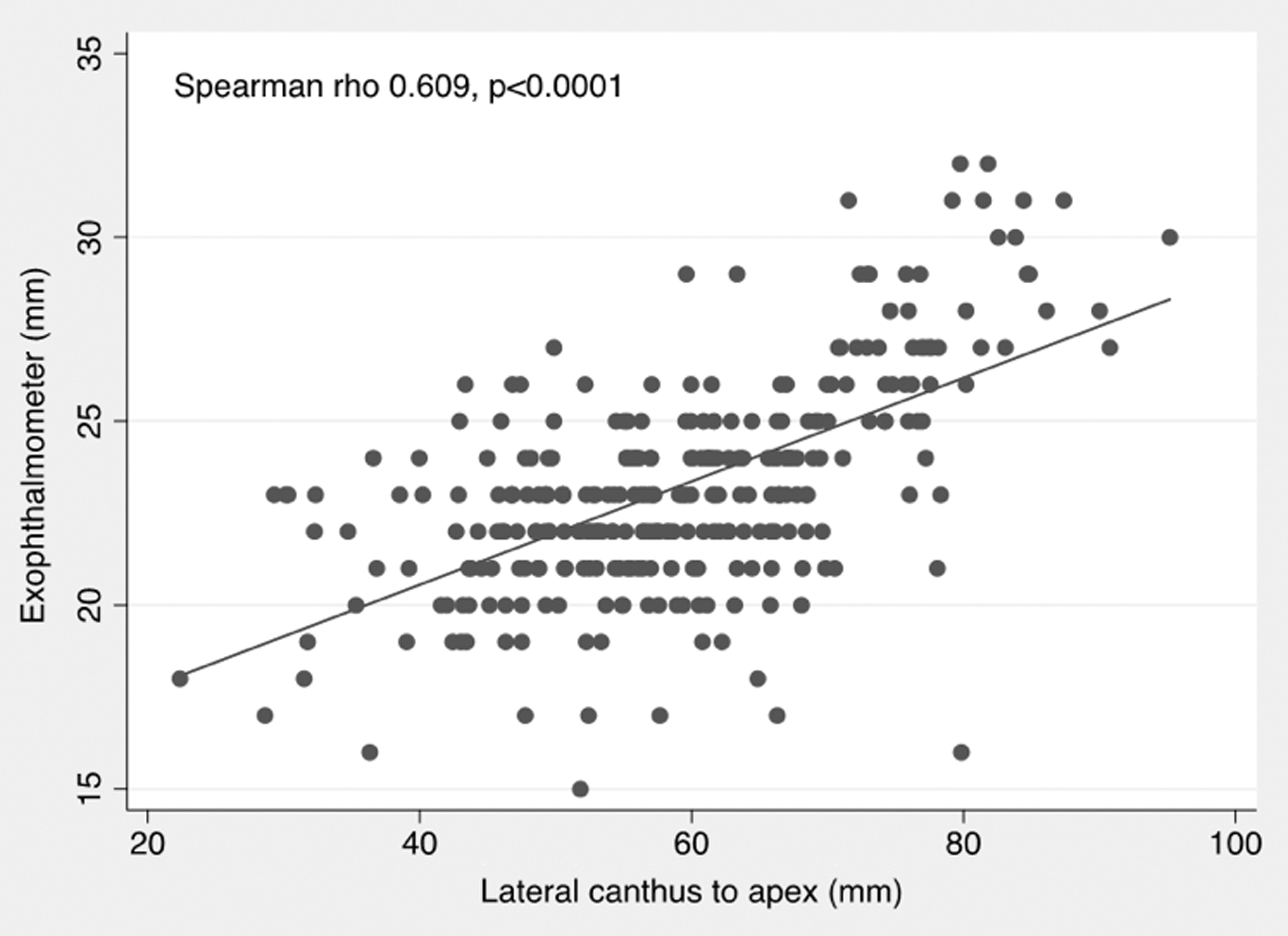
Scatter plot of clinical exophthalmometer against the LC to CA measurements by photograph, with fitted values (solid line) showing a positive correlation (Spearman's ρ = 0.609; p < 0.0001).
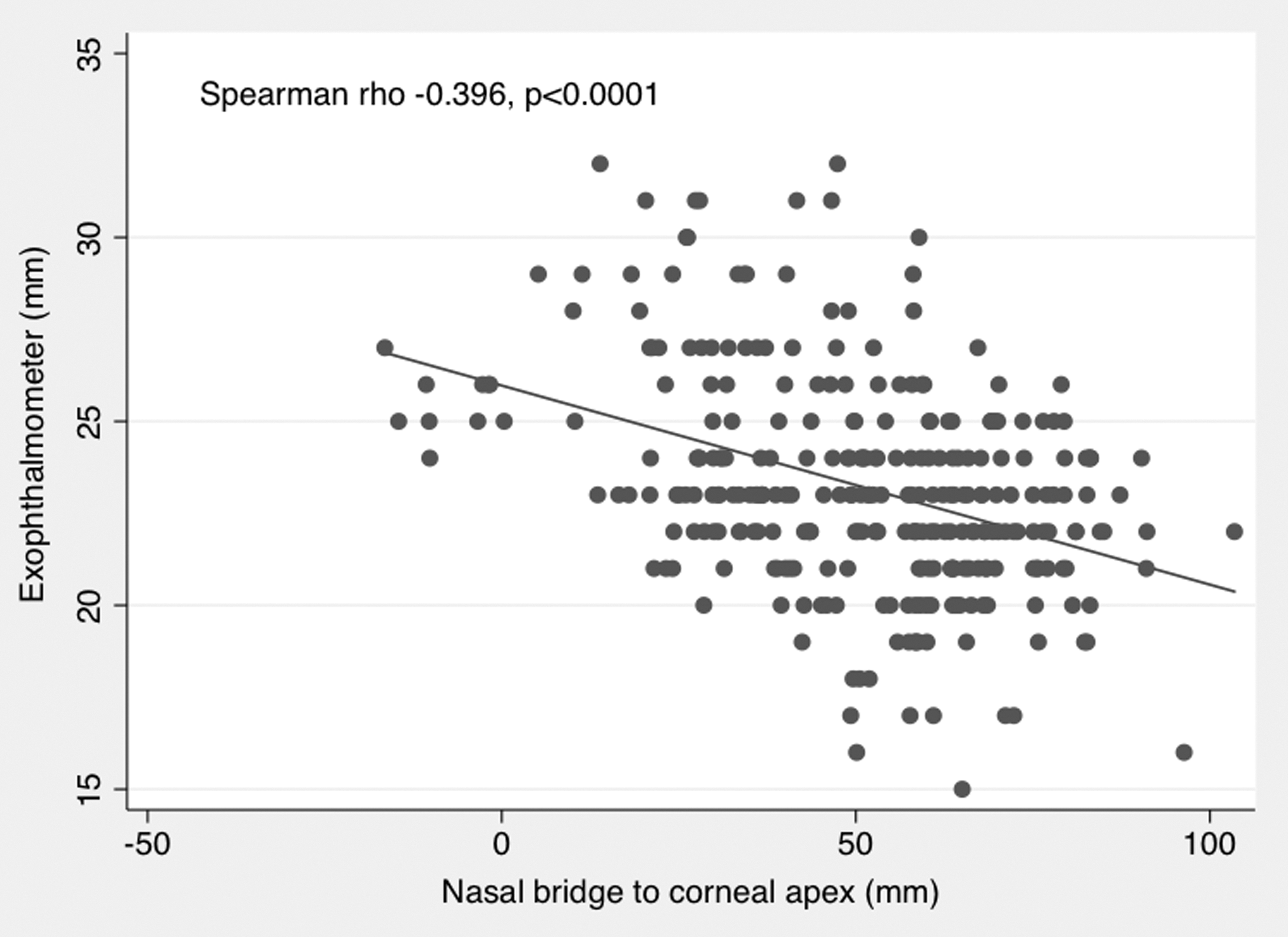
Scatter plot of clinical exophthalmometer against the NB to CA measurements by photograph, with fitted values (solid line) showing a negative correlation (Spearman's ρ = −0.396; p < 0.0001).
There was no difference between placebo and bimatoprost with regard to photo-measurement results of the lateral canthus to corneal apex distance with placebo (mean change of +1.30 mm [CI −0.74 to +3.35]) compared to bimatoprost (mean change of +0.98 mm [CI −1.25 to +3.20]; p = 0.8160). Similarly, there was no difference between placebo and bimatoprost nasal bridge to corneal apex measurement results with placebo treatment, with a mean change of −0.50 mm [CI −4.18 to +4.08] compared to bimatoprost, with a mean change of +1.30 mm ([CI −5.65 to +8.25]; p = 0.6870). There was no significant change observed in the subset of patients with unilateral proptosis (n = 12) with bimatoprost treatment, resulting in a lateral cantus measurement change of −0.32 mm [CI −4.41 to +3.76], versus untreated, resulting in a change of +1.09 mm ([CI −4.07 to +6.26]; p = 0.5252). Likewise, bimatoprost treatment resulted in a nasal bridge measurement change of +3.10 mm [CI −13.53 to +19.73] compared to the untreated eye of +6.43 mm ([95% CI −3.99 to +16.86]; p = 0.6318).
Secondary outcome analysis
In general, patients scored highly on the total visual score using the GO-QOL questionnaire throughout trial visits, with a range of mean total visual scores of 79–85. With regard to treatment, there was no change in the total visual scores. The change was calculated by subtracting post-treatment scores from baseline scores. Positive values indicated an improvement in the quality of life, and a change of at least six points was considered a minimal clinically important difference. The mean change for bimatoprost was 0.8 [CI −7.1 to 8.7] versus −0.6 for placebo ([CI −6.5 to 5.2]; p = 0.7930). There was a good negative correlation between the Gorman diplopia score and the total visual score (Spearman's ρ = −0.5118; p < 0.0001). This negative correlation persisted even after removing patients treated with prisms (Spearman's ρ = −0.5111; p < 0.0001).
Patients scored lower throughout trial visits with regard to total appearance score, with mean scores ranging from 52 to 58. No change in total appearance score was seen at three months after bimatoprost treatment, with a mean score of 0.4 [CI −3.6 to 4.5] versus 2.2 for placebo ([CI −5.2 to 9.5]; p = 0.0.6897). There was no correlation between the Gorman diplopia score and the total appearance score (Spearman's ρ = −0.0785; p = 0.3396). This correlation became significant after removing patients treated with prisms, albeit remaining a rather weak association (Spearman's ρ = −0.2282; p < 0.0115).
During trial visits, the mean IOP measured in the primary position was within the normal reference range (16–18 mmHg). As expected, bimatoprost caused a reduction in IOP, with a mean change of −2.7 mmHg [CI −4.0 to −1.4], compared to placebo, with a mean change of 0.3 mmHg ([CI −1.4 to 2.1]; p = 0.007), consistent with compliance with the medication. It was found that the chin-forward position did not alter IOP significantly. There was no difference in NHS health care expenditure between the bimatoprost period and the placebo period (Supplementary Table S2).
Bimatoprost was associated with patient-reported conjunctival hyperemia and headache (Table 3 and Supplementary Table S3). Apart from patient-reported side effects, objective assessments of photographs were also made by an independent masked assessor. Patients treated with bimatoprost had a higher detectable skin discoloration, elongation of the eyelashes, and eyelid redness compared to placebo (Supplementary Table S4). Only one (3.2%) patient developed observable periorbital fat atrophy, which was the desired effect in this trial (Fig. 6). This was a 57-year-old female patient who was a current smoker with a five-year history of GO. She was previously treated with intravenous steroids, radiotherapy, cyclosporine, and rituximab. The fat atrophy lasted for two months following the washout period. In this patient, at baseline, the right-eye exophthalmometer measurement was 23 mm, and the left-eye measurement was 24 mm. Following three months on bimatoprost, there was a reduction of 2 mm in the right eye and 1 mm in the left eye. These then returned to baseline following a washout period of two months.
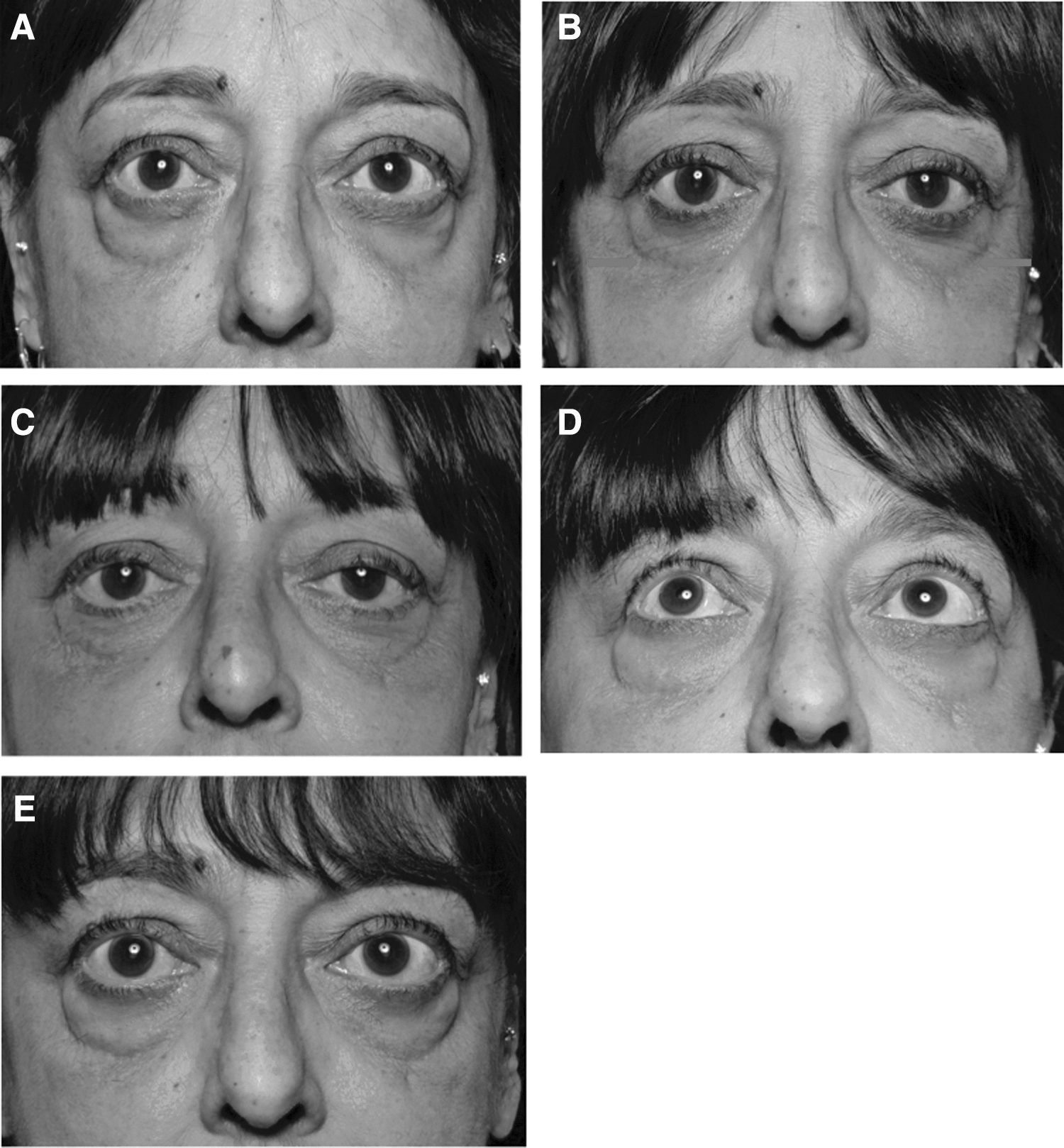
Photograph of one patient with lower-lid periorbital fat atrophy. Following three months on bimatoprost, there was a reduction of 3 mmHg in intraocular pressure in both eyes, with a 2 mm exophthalmometer reduction in the right eye and 1 mm in the left eye. These then returned to baseline following the two-month washout period. (
Patient-Reported Ocular Side Effects
The percentage was calculated from the total number of patients in the trial (N = 31).
Discussion
This is the first clinical trial to assess the effects of PGF2α in stable inactive GO. This trial did not show any clinical benefit of bimatoprost on reducing proptosis. This finding was confirmed on photographic measurements and despite the effect on IOP and appearance changes (elongation of lashes, conjunctival hyperemia, and skin changes), suggesting good compliance. The standard deviation was consistent with power calculations, suggesting that the study was not underpowered and it was unlikely that the effect was missed. This is in contrast to in vitro findings (20,21) and anecdotal case reports in people without GO (6 –8) suggesting adipocyte differentiation inhibition with bimatoprost. The findings also contrast with the results obtained with teprotumumab, a human monoclonal antibody inhibitor of IGF-IR shown to reduce proptosis. The success of teprotumumab might be attributed to the fact it was used in active GO and it targets a different pathophysiological mechanism.
There may be several explanations for the lack of effects in the primary analysis. The two main mechanisms of GO are adipogenesis and hyaluronan accumulation (22). In the “burnt out” stage, fibrosis will predominate. The topical eye drops might be absorbed less freely due to the inflammatory/fibrosis process. In the search for stable disease, in order to show the effect of PGF2α, the wrong stage of the disease might have been chosen, which is predominantly caused by hyaluronan deposition or fibrosis rather than adipogenesis. Adipogenesis starts early in the disease, and it has been shown that it may continue, even in the inactive disease stage (23). PGF2α inhibits adipogenesis per se but does not affect lipolysis and hence has no impact on an already fully mature adipocyte (20). Not all glaucoma patients treated with PGF2α develop periorbital fat atrophy, with an estimated incidence of 24.1% (24). Some patients with GO have predominantly fat excess, while the others have muscle-predominant disease (25). This suggests the possibility of a subgroup of subjects who are more susceptible to the effect of bimatoprost who could be identified by screening using orbital imaging. Perhaps a treatment duration of three months is not long enough to see the intended reduction in proptosis. However, this seems unlikely, as there was sufficient time to see fat atrophy. Compliance might also be an issue, although the changes in IOP on treatment suggest this is unlikely, and a statistically significant treatment effect was not found after adjustment was made for compliance using a reduction in IOP as a surrogate marker. Periorbital fat atrophy was observed in one (3%) subject in the patient population, suggesting that periorbital fat atrophy is different from general fat reduction. Perhaps the periorbital effect seen is mediated via a different mechanism such as activation on matrix metalloproteinases (26).
The assessment of exophthalmos was robust with an exophthalmometer and supported with photographic assessments conducted by an assessor who was masked to the treatment phase. There were two trained assessors in the current study. Assessors were assigned to the trial patients at each trial visit according to each assessor's availability. To reduce inter-rater variation, the assessors were calibrated by multiple exophthalmometer readings on the same non-trial subjects in the clinic, and adjustments were made to ensure their readings were comparable. Photographic measurements also provided further independent confirmation of the exophthalmometer results.
The strengths of this trial include its crossover design, with no period or carry-over effects. There was good patient retention and good compliance, as evidenced by the fall in IOP in the treatment phase. The success of the masking process was analyzed by asking patients and assessors directly and by the independent masked assessor on photographic assessment. Assessors guessed treatment allocation incorrectly in 56.7% of the patients. Approximately 27% of the patients on placebo thought that the prominence of their eyes improved compared to 43% treated with bimatoprost. A little more than 40% of the patients in both phases preferred the treatment. Forty-three percent of subjects in the placebo phase were unsure of treatment allocation, and a further 10% guessed incorrectly; 29% in the bimatoprost phase were unsure, and 32% guessed incorrectly when asked about their treatment allocation, suggesting that masking was successful.
In summary, bimatoprost treatment over three months in inactive GO did not result in improvements in proptosis, and this information should prevent clinicians trialing this approach further and causing side effects unnecessarily. Future trials should be done on early-stage GO and active disease. Periorbital fat atrophy appears to be an idiosyncratic reaction to bimatoprost rather than a routine event in inactive GO patients. The BIMA study has demonstrated that crossover studies can be performed reliably in patients with persistent proptosis due to thyroid eye disease and that this study design is acceptable to patients. The BIMA study also has shown that >60% of patients with residual proptosis in thyroid eye disease also have double vision (diplopia). IGF-1R antagonists have shown promise in active disease, but still only surgical treatments are available in “burnt out” disease. Hence, there are still large unmet needs in this patient group.
Footnotes
Acknowledgments
Funding was provided by Health and Care Research Wales under the Research for Patient and Public Benefit Wales Scheme.
Author Disclosure Statement
No competing financial interests exist for any author.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Data—BIMA study protocol
Supplementary Data—Standard operating procedure