
Abstract
Background:
Vemurafenib is a selective BRAF inhibitor (BRAFi) that has shown promising activity in BRAFV600E -positive papillary thyroid cancer (PTC). However, adverse events and resistance to a single-agent BRAFi often require discontinuation of the targeted therapy in BRAFV600E -positive PTC. Thus, this study investigated the expression of anti-apoptotic B-cell lymphoma 2 (BCL-2) family members, which are frequently overexpressed in many human cancers to inhibit apoptosis, in PTC harboring the BRAFV600E mutation after BRAFi treatment, and then evaluated the cytotoxic effects of a homology 3 domain (BH3)-mimetic in combination with a BRAFi.
Methods:
K1 cells (BRAFV600E -positive human PTC) were treated with various concentrations of vemurafenib to investigate the effect of the BRAFi. In addition, the study analyzed the protein expression profiles of phosphorylated ERK1/2 (p-ERK 1/2) and anti-apoptotic BCL-2 family after vemurafenib treatment and selected the target anti-apoptotic protein. Antitumor effects were measured by cell counting, and effects on apoptosis were determined by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay and Western blot analysis.
Results:
At a concentration of 10 μM, vemurafenib inhibited the growth of K1 cells by 49.4%. Western blot analysis following exposure to 10 μM vemurafenib revealed that p-ERK1/2 gradually decreased over 24 hours, but the expression of B-cell lymphoma-extralarge (BCL-XL) and BCL-2 increased after 12 hours of treatment. Based on this result, the K1 cells were treated with navitoclax (BCL-2/BCL-XL inhibitor) for 24 hours up to a concentration of 4 μM, which resulted in negligible effects on cell survival. However, a combination treatment of 0.5 μM navitoclax with 1 μM vemurafenib resulted in significantly enhanced cell growth inhibition and increased apoptosis.
Conclusions:
The results of the present study show that vemurafenib increased the expression of anti-apoptotic proteins of the BCL-2 family. Thus, the combination of vemurafenib with navitoclax may be effective in BRAFV600E -positive PTC treatment.
Introduction
Although the majority of patients with papillary thyroid cancer (PTC) have excellent long-term outcomes with standard surgical treatment and radioiodine therapy, a significant number suffer from recurrences. Some patients have an aggressive course and present with locally advanced disease or distant metastasis, and there is currently no curative treatment if the cancer becomes surgically inoperable and/or loses radioiodine avidity. The BRAFV600E mutation strongly correlates with aggressive tumor characteristics such as metastasis, disease recurrence, and a loss of radioiodine avidity resulting in failure of radioiodine therapy (1). Thus, thyroid carcinomas harboring the BRAFV600E mutation are more likely to require systemic therapy for unresectable recurrent or metastatic diseases.
The BRAFV600E mutation is a major cause of aberrant activation of the MAPK pathway, which plays an essential role in mediating enhanced cellular proliferation and in cellular survival. Mutation in BRAF, the most frequent genetic alteration in thyroid cancer, is known to be present in approximately 44% (38–83% depending on the studies) of PTC and in 78% of recurrent PTC (1 –3). Therefore, the BRAFV600E mutation is a good therapeutic target, and the development of BRAF inhibitors (BRAFi) is attracting great attention in the pharmaceutical industry. Vemurafenib (PLX4032) is the first orally available selective BRAFi approved by the Food and Drug Administration for metastatic melanoma with the BRAFV600E mutation, and it has shown high treatment efficacy for malignant melanoma (4). Several clinical studies have suggested that vemurafenib also shows promising activity in BRAFV600E -positive PTC (5,6). However, resistance to single-agent BRAFi often results in failure of the targeted therapy in BRAFV600E -positive PTC (5,7) as well as in melanoma (8,9). One strategy to delay the onset of resistance is to formulate a suitable drug combination to overcome the resistance.
A potential combination of targets are the anti-apoptotic proteins of the B-cell lymphoma 2 (BCL-2) family, such as BCL-2, B-cell lymphoma-extralarge (BCL-XL), and myeloid cell leukemia-1 (MCL-1), which are frequently overexpressed in many human cancers and protect from apoptosis (10). Overexpression of the anti-apoptotic proteins contributes to apoptosis resistance in multiple types of cancer, and therefore the anti-apoptotic proteins could be important targets. On the contrary, pro-apoptotic proteins, BCL-2-antagonist/killer (BAK) and BCL-2-associated X (BAX), facilitate apoptosis through activation of the mitochondrial cell death pathway. Activation of BAK and BAX is initiated through interactions with the BCL-2 homology 3 domain (BH3)-only proteins, BID and BIM (11). Small-molecule BH3 mimetics, such as navitoclax (ABT-263), disrupt BCL-2/BCL-XL interactions with pro-apoptotic proteins such as BIM, thereby inducing apoptosis (12). Proteins of the BCL-2 family play key roles in the regulation of apoptosis in chemotherapeutic strategies.
The present study investigated the expression of anti-apoptotic BCL-2 family members in PTC harboring the BRAFV600E mutation after BRAFi treatment. In addition, it evaluated the cytotoxic effects of combining a BH3-mimetic and a BRAFi in a BRAFV600E -positive PTC cell line.
Methods
Cell culture and compounds
The human BRAFV600E -positive PTC cell line K1 was purchased from Sigma–Aldrich (St. Louis, MO), and the short tandem repeat (STR) profile is identical to the one documented on the Sigma–Aldrich Web site. K1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/Ham's F12 50/50 media (Corning Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (FBS; Gibco, Rockville, MD) and 1% penicillin-streptomycin (Hyclone, Logan, UT) in a humidified incubator at 37°C and 5% CO2. Vemurafenib (PLX4032, BRAFi) and navitoclax (ABT-263, BCL-2/BCL-XL inhibitor) were purchased from Selleck Chemicals (Houston, TX). Stock solutions of each inhibitor were dissolved in dimethyl sulfoxide (Sigma–Aldrich) and stored at −20°C.
Cytotoxicity assay
Survival curves of the K1 cells were assessed using a cell counting kit-8 assay (Dojindo Molecular Technologies, Rockville, MD). Briefly, 5 × 103 cells per well were seeded onto 96-well plates containing the DMEM-F12 medium supplemented with 2.5% FBS, and the chemicals were treated at suitable concentrations for 24 h. Following incubation with the chemicals, CCK-8 reagents were added to the wells, and absorbance was measured at 450 nm using a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA). Cell viability results were expressed as a percentage of that of vehicle.
FACS analysis
For cell-cycle analysis, K1 cells were treated with different concentrations of vemurafenib for 24 hours. Cell pellets were washed in phosphate-buffered saline, fixed overnight with 70% ethanol, treated with 250 μg/mL RNase A (Sigma–Aldrich), and then stained with 50 μg/mL propidium iodide (PI) solution (BD Biosciences, San Jose, CA). DNA staining with PI was analyzed by florescence activated cell sorting (FACS Calibur™; BD Biosciences, San Diego, CA).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay
For the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay in vitro, K1 cells were seeded at a density of 2 × 104 and treated with 0.5 μM navitoclax, 1 μM vemurafenib, and 0.5 μM navitoclax plus 1 μM vemurafenib for 24 hour in a four-well chamber slide. Apoptotic K1 cells were detected by the TUNEL assay using the DeadEnd™ Fluorometric TUNEL system (Promega, Fitchburg, WI) according to the manufacturer's protocol. The samples were covered with VECTASHIELD® mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). TUNEL-positive (green) and DAPI-positive cells (blue nuclear stain) were observed by confocal laser microscopy (LSM 5 Exciter; Carl Zeiss, Oberkochen, Germany).
Protein isolation and Western blot analysis
The K1 cells were cultured with chemicals for 24 hours, and total protein was isolated using radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, Rockford, IL) containing a protease and phosphatase inhibitor cocktail kit (Thermo Fisher Scientific). The lysed cells were vortexed three times and centrifuged at 13,000 g at 4°C. Then, the supernatants were collected and quantified with the bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Membrane and cytosolic proteins were extracted using a Mem-PER™ Plus Membrane Protein Kit (Thermo Fisher Scientific). The collected cell pellets were washed with cell wash solution and centrifuged. After discarding the supernatant, permeabilization buffer was added to the cell pellets. The cell pellets were centrifuged, and the cytosolic proteins were extracted in the supernatant. Solubilization buffer was added to the remnant pellets, which were centrifuged to extract the membrane proteins.
Equal amounts of proteins were loaded in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA). The membranes were blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline containing Tween-20 (TBS-T) for one hour and probed with the respective primary antibodies in 0.5% BSA overnight at 4°C. Membranes were probed with horseradish peroxidase (HRP)-conjugated secondary antibodies for one hour at room temperature after washing with TBS-T. The signals were visualized using ECL detection reagent (GE Healthcare Life Sciences, Milan, Italy), and bands were detected on the Fusion FX chemiluminescence analyzer system (Vilber Lourmat, Collégien, France).
The primary antibodies used were: phospho-p44/42 MAPK (p-ERK1/2), p44/42 MAPK (total ERK1/2), BCL-2, BCL-XL, MCL-1, BAX, BIM, caspase-3, and cleaved-caspase 3. Cleaved-PARP antibodies were purchased from Cell Signaling Technology (Danvers, MA); cytochrome C antibody was acquired from Abcam (Cambridge, United Kingdom); luciferase antibody was purchased from Promega (Madison, WI); and GAPDH and β-actin antibodies were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). The following HRP-conjugated secondary antibodies were used: anti-goat (Santa Cruz Biotechnology) or anti-rabbit (Cell Signaling Technology).
Statistical analysis
All data are expressed as the mean ± standard deviation, and two groups of data were statistically analyzed by Student's t-test using GraphPad Prism v5.01 (GraphPad Software, Inc., San Diego, CA). A p-value <0.05 was considered statistically significant. Error bars represent standard deviations.
Results
Anti-proliferative effect of vemurafenib in K1 cells
To investigate the anti-proliferative effect of the BRAFi vemurafenib on the BRAFV600E -positive PTC cell line, K1 cells were treated with various concentrations of vemurafenib for 24 hours, and then the percentage of viable cells compared to those treated with vehicle was assessed. As seen in Figure 1A, at a concentration of 10 μM, vemurafenib inhibited the growth of K1 cells by 49.4%. However, cell-cycle analyses showed a marked G1 arrest at 24 hours. There was no significant change in the percentage of cells in the subG1 phase as the concentration of vemurafenib increased (Fig. 1B).
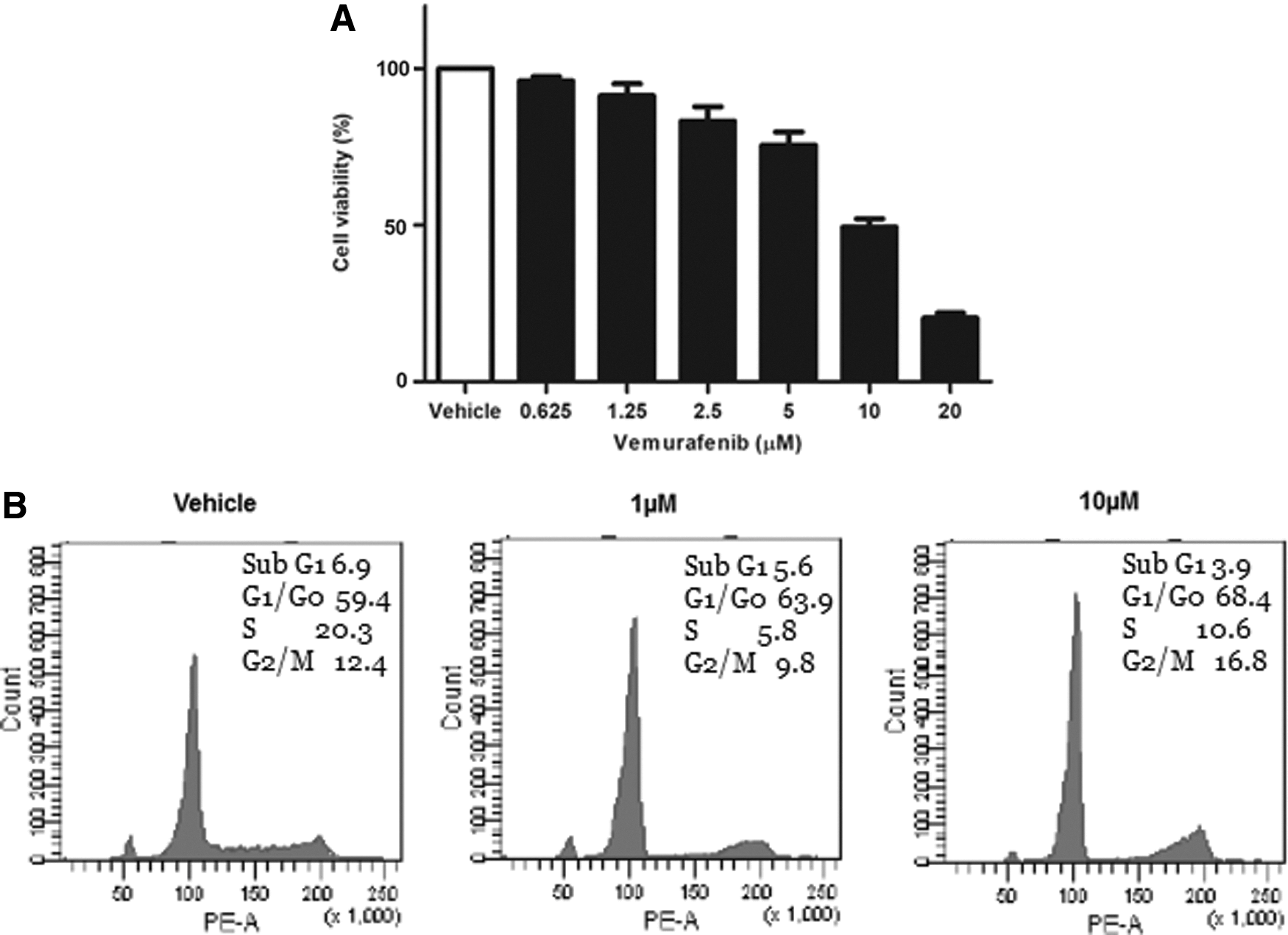
The effect of vemurafenib in K1 cells. (
Vemurafenib treatment induces BCL-2 and BCL-XL expression
The protein expression profiles of p-ERK1/2 and pro-survival BCL-2 family were analyzed following 1, 6, 12, 24, and 48 hours of exposure to 10 μM vemurafenib. Phosphorylated ERK1/2 gradually dropped over 24 hours but rebounded at 48 hours of treatment. A sharp reduction of MCL-1 was observed after six hours, but the expression of BCL-2 and BCL-XL increased by 3.6- and 1.9-fold, respectively, after 24 hours of vemurafenib treatment. The increase in BCL-2 and BCL-XL expression was induced by vemurafenib treatment (Fig. 2).
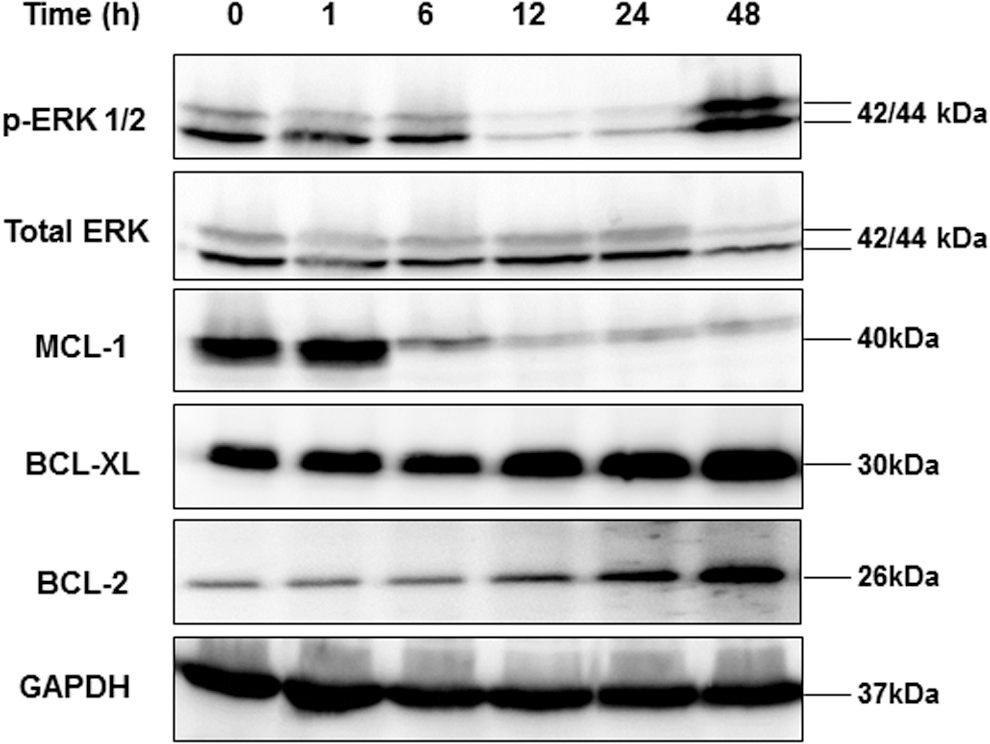
Vemurafenib treatment induces B-cell lymphoma 2 (BCL-2) and B-cell lymphoma-extralarge (BCL-XL) expression. Western blot analysis of phosphorylated ERK1/2 and anti-apoptotic BCL-2 family in the K1 cells following 1, 6, 12, 24, and 48 hours of exposure to 10 μM vemurafenib. Phosphorylated ERK1/2 gradually dropped over 24 hours but rebounded at 48 hours of treatment. The expression of BCL-XL and BCL-2 was increased after 12 hours of treatment. A sharp reduction in myeloid cell leukemia-1 was observed after six hours of treatment. GAPDH was used as a loading control.
Synergistic effect of vemurafenib and navitoclax
Since the cells that survived after vemurafenib treatment had upregulation of BCL-2 and BCL-XL, the study then evaluated whether navitoclax, which is a potent inhibitor of BCL-XL and BCL-2, enhances the vemurafenib-mediated cytotoxic effect. Navitoclax alone exhibited negligible effects on the survival of K1 cells up to a concentration of 4 μM (Fig. 3). However, the cytotoxic effect of a combination treatment was significantly higher than each individual drug treatment. The combination of vemurafenib (1 μM) and navitoclax (0.5 μM) reduced cell survival by 39.4%, whereas the percentages of cells that survived with individual drug treatments were 90.6% after the 1 μM vemurafenib treatment and 99.3% after the 0.5 μM navitoclax treatment (Fig. 4A). Furthermore, K1 cells treated with the combination of vemurafenib and navitoclax showed significant TUNEL staining, indicating apoptosis, compared to those treated with each individual drug (Fig. 4B). To confirm the cell apoptosis and to understand the relationship better between cellular responses to BRAFi and the BCL-2 family proteins, the study also assessed the mitochondrial pathway of apoptosis as a target downstream of the vemurafenib treatment by Western blot analysis. As shown in Figure 5A, vemurafenib treatment did not induce an appreciable change of pro-apoptotic BAX but promoted a marked expression of pro-apoptotic BIM. However, anti-apoptotic BCL-XL and BCL-2 were also increased by 1.7- (Fig. 5B) and 1.9-fold (Fig. 5C), respectively, following vemurafenib treatment. The levels of caspase-3, cleaved caspase-3, cleaved PARP in total cell lysates, and cytochrome-c in cytosolic protein were examined. The expressions of cleaved caspase-3, cleaved PARP, and cytochrome-c were increased by 3.3-, 9.1-, and 2.2-fold, respectively, after the combination treatment for 24 hours (Fig. 5D).
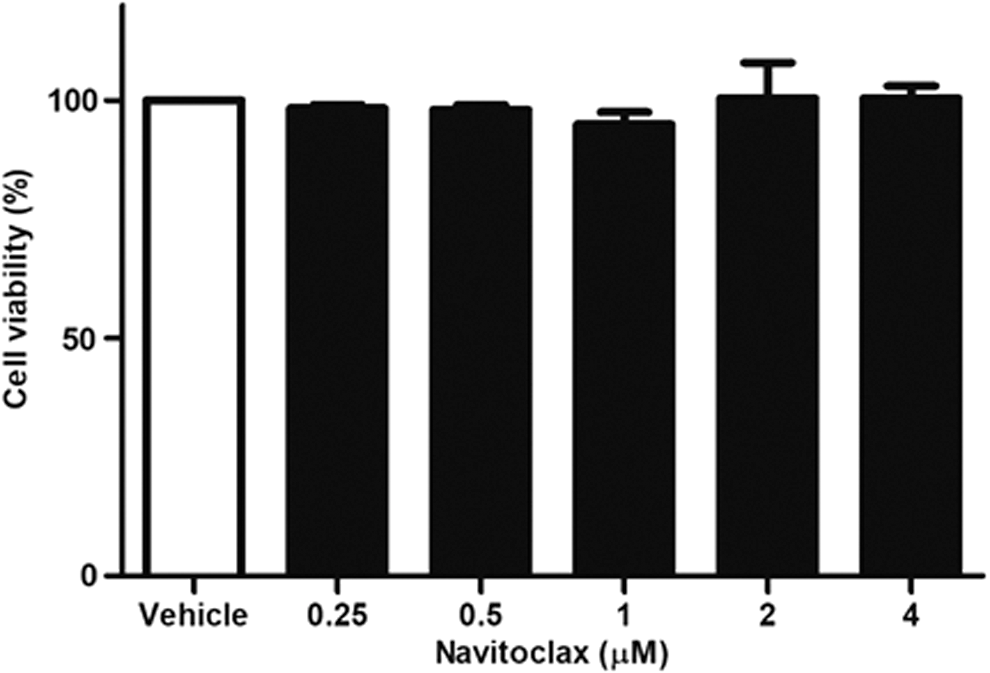
Cell viability of K1 cells after navitoclax treatment. K1 cells were treated with various concentrations of navitoclax (BCL-2/BCL-XL inhibitor, ABT-263) for 24 hours. Navitoclax alone, up to a concentration of 4 μM, exhibited negligible effects on the survival of K1 cells.
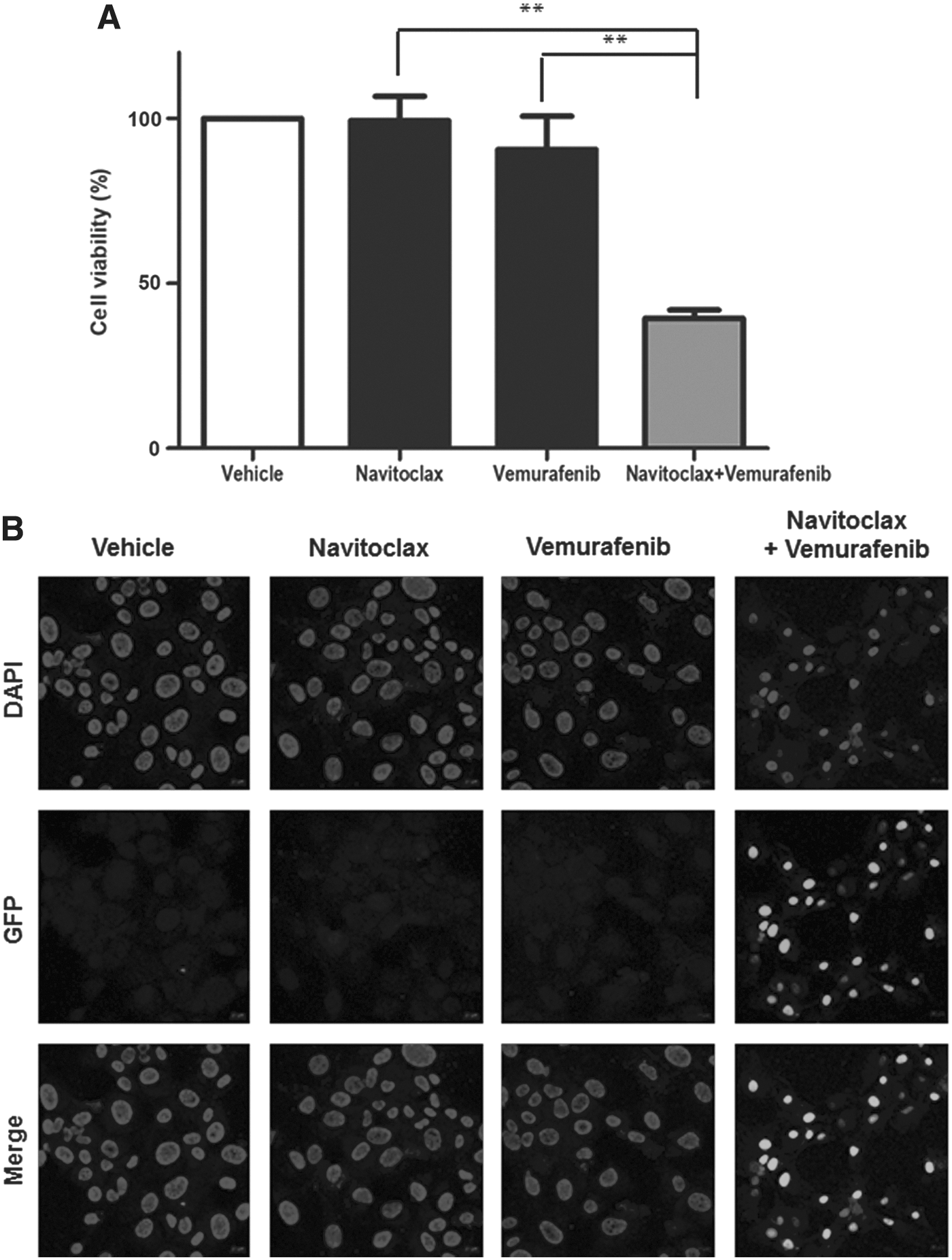
Combination treatment of navitoclax and vemurafenib in K1 cells. The combination treatment of 0.5 μM navitoclax with 1 μM vemurafenib showed 39.4% cell viability (

Western blot analysis of the BCL-2 family proteins in the combination treatment. (
Discussion
Therapeutic application of BRAFi offers a new strategy in the treatment of metastatic radioiodine-refractory thyroid cancers expressing the BRAFV600E mutation. In a recent Phase II trial, the clinical responses to vemurafenib were encouraging in both groups, regardless of whether they received a multi-kinase inhibitor (cohort 2) or not (cohort 1). However, the beneficial effects were not sustained, as the median time to progression was 18.2 months in cohort 1 and 8.9 months in cohort 2 (6).
One confirmed mechanism of resistance to BRAFi is the inherent negative feedback mechanism of the MAPK pathway. In BRAFV600E -mutated tumors, BRAFV600E activation is RAS independent, and receptor tyrosine kinases (RTK) signaling is suppressed because high levels of ERK induce strong ERK-dependent negative feedback. Thus, at baseline, these tumors are relatively insensitive to the effects of secreted growth factors. In addition, BRAF is primarily in a monomeric and drug-sensitive state in this setting. However, inhibition of RAF monomers and ERK signaling by vemurafenib causes a relief of ERK-dependent feedback, and growth factors can signal to reactivate RTK signaling to RAS. With RAS activation, RAF kinases dimerize, which allows vemurafenib to induce trans-activation of RAF. The RAF dimers are resistant to BRAFi, thereby bypassing the inhibition and reactivation of MEK and then ERK (13). The rebound of ERK activity can attenuate the antitumor effect of BRAFi treatment, and this feedback mechanism in the MAPK pathway makes a complete response of disease unusual (6). Thyroid cancer cells show rapid increases in RAS-GTP and rebound in p-ERK after BRAFi treatment, consistent with the activation of upstream signals (14). The present study also demonstrates reactivation of p-ERK after 48 hours of vemurafenib treatment (Fig. 2). A previous study reported that ERK reactivation in response to BRAFi induces HER3 gene expression by releasing transcription repressor CTBP protein from the HER3 promoter. Autocrine-secreted neuregulin-1 (NRG1) binds to HER3 and triggers HER3/HER2 heterodimerization and receptor phosphorylation, thereby increasing the activation of the MAPK pathways (14). Several other resistance mechanisms for BRAFi have been demonstrated in thyroid cancer such as reactivation of the MAPK pathway through alternate BRAF splicing (15) or KRASG12D mutation (16), activation of the PI3K/AKT pathway through c-MET (17), and autocrine interleukin-6-mediated JAK/STAT3 and MAPK signaling induction (18).
Moreover, inhibition of the ERK signaling pathway by BRAFi causes only G1 cell-cycle arrest, inducing minimal apoptosis in thyroid cancer cells. After vemurafenib treatment, a small proportion of K1 cells were in the subG1 phase, with an increase in the G1 phase (Fig. 1B), suggesting that the treatment is cytostatic rather than cytotoxic. This result is consistent with recently published BRAFi data on thyroid cancer cell lines (19,20). In this regard, although inhibition of the MAPK pathway by BRAFi induces G1 cell-cycle arrest in thyroid cancer cells, the ERK pathway could be activated constitutively by the mechanisms described above. This may provide opportunities to remodel the ERK1/2 pathway, which facilitates resistance to BRAFi.
Apoptosis can be initiated by various intracellular stresses and is elicited by most chemotherapy agents through intrinsic and extrinsic pathways. However, BRAFi fails to induce apoptosis in thyroid cancer. Thus, this study evaluated the expression of BCL-2 family proteins, which have been suggested to be key to confer resistance to apoptosis and promote survival. The members of the BCL-2 family are mainly involved in the intrinsic pathway by regulating mitochondrial outer membrane permeabilization (MOMP). MOMP causes the release of cytochrome c from the mitochondrial intermembrane space into the cytoplasm, which leads to activation of caspases and cell death. The BCL-2 family is composed of two types: pro-apoptotic and anti-apoptotic proteins. The pro-apoptotic members are divided into “effectors” and “BH3-only proteins.” The effector proteins BAK and BAX oligomerize to form pores releasing cytochrome c. BAK/BAX activation is triggered by the “direct activator” BH3-only proteins BID and BIM. Various stress stimuli activate BH3-only proteins. Anti-apoptotic proteins such as BCL-2, BCL-XL, and MCL-1 preserve survival by binding and sequestering activator BH3-only proteins and activated BAK/BAX. However, the other BH3-only proteins such as BAD and PUMA, described as “sensitizer” BH3-only proteins, can displace activator BH3 proteins or activated BAK/BAX from anti-apoptotic proteins (11,21,22). The balance between pro-apoptotic and anti-apoptotic signals decides the fate of each cell.
In the present study, vemurafenib treatment led to an increase in the pro-apoptotic BIM protein (Fig. 5A). BIM, when phosphorylated by ERK1/2, is rapidly degraded via the proteasome pathway (23). Thus, hyperactivation of MAP kinase signaling may allow the cancer cell to suppress BIM protein levels and evade apoptosis. BRAFi may prevent phosphorylation of BIM by ERK, thereby facilitating increased BIM expression and apoptosis (Fig. 6). However, vemurafenib alone did not change the expression of cytosolic cytochrome c, cleaved caspase-3, and cleaved PARP compared to vehicle, which suggests a failure to induce apoptotic signaling because the levels of anti-apoptotic BCL-XL and BCL-2 were also increased by vemurafenib treatment. Pro-apoptotic signaling arises due to increased expression of BIM proteins, but it was suppressed by binding with anti-apoptotic BCL-2 proteins. It is unclear how BRAFi mediates increased expression of the anti-apoptotic proteins BCL-XL and BCL-2. In a previous study using patient tumor samples with metastatic melanoma positive for the BRAFV600E mutation, BRAFi therapy induced an increase in the mRNA levels of anti-apoptotic BCL-2 family members, although these mRNA changes did not translate into changes of protein expression (24). However, the expression of the anti-apoptotic proteins BCL-XL and BCL-2 increased in response to BRAFi therapy in the present study using a BRAFV600E -positive PTC cell line. Expression of anti-apoptotic proteins can be regulated at both the transcriptional and post-translational levels by BRAFi, and future studies are needed to elucidate the exact mechanism of the effect.
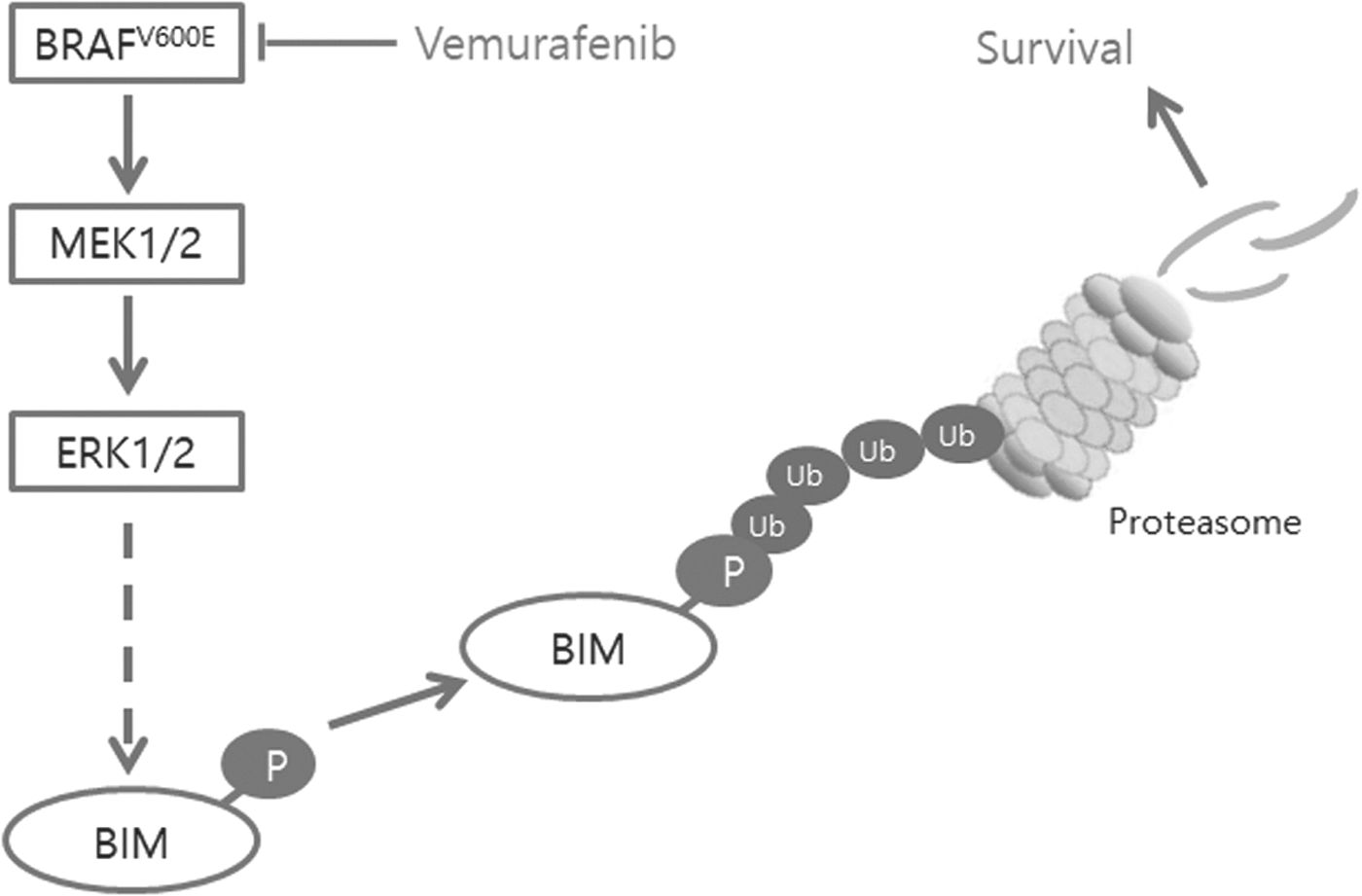
Cancers with the BRAFV600E mutation result in constitutive activation of the MAPK pathway, and phosphorylation of BIM by ERK induces its degradation by the proteasome pathway and promotes cell survival. Treatment with vemurafenib, a BRAF inhibitor, blocks these pathways and leads to upregulation of proapoptotic BIM protein, thereby inducing apoptosis.
The addition of navitoclax, a BCL-XL and BCL-2 inhibitor, reduced the expression of BCL-XL and BCL-2, resulting in cytochrome c release and a marked increase of cleaved caspase-3 and cleaved PARP. These results suggest that the majority of pro-apoptotic BIM induced by vemurafenib was silenced by the anti-apoptotic BCL-2 family, thereby ensuring cell survival. Navitoclax can promote vemurafenib-mediated apoptosis, probably by releasing pro-apoptotic BIM from anti-apoptotic proteins. Thus, a combination treatment of vemurafenib and navitoclax could move cytostatic responses to cell death and would have the added effect of increasing antitumor activity. Although just a single human BRAFV600E -positive PTC cell line was used in this study, another study used BRAF-mutant melanoma cell lines. Fofaria et al. showed overexpression of anti-apoptotic proteins of the BCL-2 family in response to BRAFi treatment in BRAF-mutant melanoma cell lines (25). The present study also demonstrates that the BRAFi treatment in BRAF-mutant thyroid cancer cell line increases the expression of the anti-apoptotic proteins of the BCL-2 family, and shows promising results of the combination treatment with a BRAFi and a BH3 mimetic for BRAFV600E -positive PTC. Moreover, in the present study, the combination treatment of vemurafenib and navitoclax has an anti-proliferation effect and induces apoptosis even at lower concentrations compared to treatments with the individual agents. In a previous study, BRAF-mutant melanoma cell lines were uniformly sensitive to growth inhibition by vemurafenib (IC50 <100 nM), whereas most thyroid and colorectal cancer cell lines were comparatively refractory (IC50 >1 μM) (14). Thus, this combination may decrease the frequency and severity of adverse effects of the drugs, since the concentrations required to achieve a durable response will likely be lower.
Conclusion
The results of the present study demonstrate that vemurafenib alone has a transient effect due to the reactivation of the MAPK pathway and overexpression of members of the anti-apoptotic BCL-2 family in BRAFV600E -positive PTC, which limits long-term therapeutic efficacy. However, the combination of vemurafenib and navitoclax can induce substantial cell death and exert cytotoxic effects in BRAFV600E -positive PTC. Moreover, the combination of vemurafenib and navitoclax requires lower doses to obtain a therapeutic effect compared to doses with single drug treatments. Therefore, it may be applied as a safer and efficient therapeutic strategy in BRAFV600E -positive PTC.
Footnotes
Acknowledgments
This work was supported by Biomedical Research Institute grant, Kyungpook National University Hospital (2016).
Author Disclosure Statement
The authors have nothing to disclose and claim that no competing financial interests exist.