
Abstract
Background:
Loss of the expression of thyroid differentiation markers such as sodium iodide symporter (NIS) and, consequently, radioiodine refractoriness is observed in aggressive papillary thyroid cancer and anaplastic thyroid cancer (ATC) that may harbor the BRAFV600E mutation. Activation of the BRAFV600E oncogene in thyroid follicular cells induces the expression of the miR-17-92 cluster that comprises seven mature microRNAs (miRNAs). miRNAs are a class of endogenous small RNAs (∼22 nt) that regulate gene expression post-transcriptionally. Indeed, miR-17-92 is overexpressed in ATC, and in silico prediction shows the potential targeting of thyroid transcription factors and tumor suppressor pathways. In this study, we aimed to investigate the role of the miR-17-92 cluster in thyroid cell differentiation and function.
Methods:
miR-17-92 silencing was performed using CRISPR/Cas9n-guided genomic editing of the miR-17-92 gene in the KTC2 ATC cell line, and miR-17-92 cluster or individual miRNAs were overexpressed in PCCl3 thyroid cells to evaluate the influence in thyroid cell differentiation and cell function.
Results:
In this study, we demonstrate that CRISPR/Cas9n gene editing of the miR-17-92 cluster results in promotion of thyroid follicular cell differentiation (NIS, thyroperoxidase, thyroglobulin, PAX8, and NKX2-1 expression) in the KTC2 ATC cell line and inhibits cell migration and proliferation by restoring transforming growth factor beta (TGF-β) signaling pathway responsiveness. Moreover, induction of the miR-17-92 cluster in normal thyroid follicular cells strongly impairs thyroid differentiation and induces a pro-oncogenic effect by blocking TGF-β signaling and increasing cell migration.
Conclusions:
miR-17-92 is a potent regulator of thyroid follicular cell differentiation, and CRISPR/Cas9n-mediated editing of the miR-17-92 gene efficiently blocks miR-17-92 expression in the KTC2 ATC cell line, resulting in improvement of thyroid differentiation. Thus, targeting miR-17-92 could provide a potential molecular approach to restoring thyroid cell differentiation and NIS expression in aggressive thyroid cancer.
Introduction
Thyroid cancer is the most frequent endocrine cancer and the most rapidly increasing cancer in incidence (1). Thyroid oncogenesis is mediated mainly by genetic alterations in MAPK signaling, with RET/papillary thyroid cancer (PTC) rearrangements, as well as RAS and BRAF mutations as the most prevalent alterations in PTC (2,3). Anaplastic thyroid cancer (ATC) is the most aggressive histotype of thyroid malignancy, and its pathogenesis remains incompletely understood. High-throughput next-generation sequencing data support a model of multistep carcinogenesis with ATC developing from well-differentiated tumors (4), and the main genetic alterations detected in ATC comprise TP53, BRAF, and TERT mutations (5 –8). Nevertheless, most ATC cases are fatal due to the rapidly progressing tumors that become unresectable, refractory to radioiodine therapy, and that develop metastatic disease. Remarkably, recent studies described impressive results in the treatment of a BRAFV600E advanced ATC with a neoadjuvant combined therapy of BRAF + MEK inhibitors (dabrafenib + trametinib) and immunotherapy that turned the tumor resectable, and treatable with chemoradiation and sustained control with BRAF therapy in combination with immunotherapy (9,10). Indeed, the combined therapy of dabrafenib + trametinib has been recently approved by the Food and Drug Administration for the treatment of patients with locally advanced or metastatic ATC positive for the BRAFV600E mutation and with no satisfactory locoregional treatment options.
MicroRNAs (miRNAs), small noncoding RNAs (∼22 nt), have been recognized as potent regulators of gene expression; they target post-transcriptional regulation of messenger RNAs (mRNAs), modulating the vast majority of processes that contribute to tumorigenesis and progression. miRNA deregulation is one of the hallmarks of cancer (11), and high levels of the miR-17-92 cluster have been detected in ATC (12). The miR-17-92 cluster, also known as oncomiR-1, transcribes seven mature miRNAs (miR-17-5p, miR-17-3p, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92a) that ultimately exert a pro-oncogenic effect in various types of cancers (13 –15). High-throughput miRNA expression studies have shown increased expression of miR-17-92 in PTC (16), and our group showed that BRAFV600E activates the miR-17-92 cluster in normal thyroid follicular cells (17). The BRAFV600E oncogene is associated with a loss of expression of the sodium iodide symporter (NIS/SLC5A5) and induces genomic instability in normal thyroid follicular cells (18), thus contributing to refractoriness to radioiodine therapy and disease progression, as observed in the aggressive subtypes of PTC and ATC (19). An in silico search showed that miRNAs can potentially target genes involved in thyroid cell differentiation (e.g., PAX8) and various components involved in thyroid hormone synthesis including NIS and thyroperoxidase (TPO) (20).
Investigating the role of miR-17-92 could improve the understanding of thyroid cancer aggressiveness due to loss of differentiation. In this study, we demonstrate that CRISPR/Cas9n gene editing of the miR-17-92 cluster results in promotion of thyroid follicular cell differentiation by increasing the expression of NIS, TPO, thyroglobulin (TG), PAX8, and NKX2-1 (previously known as TTF1) in the KTC2 ATC cell line, while inhibiting cell migration and proliferation by restoring transforming growth factor beta (TGF-β) signaling pathway responsiveness. Moreover, induction of the miR-17-92 cluster in normal thyroid follicular cells strongly impaired thyroid differentiation and induced a pro-oncogenic effect. Thus, miR-17-92 plays an important role in thyroid biology and differentiation, and modulating this cluster of miRNAs could improve thyroid cancer differentiation and cancer management.
Methods
Cell lines and culture
The human ATC cell line KTC2 was cultivated in RPMI supplemented with 5% fetal bovine serum (FBS). Stable KTC2 clones with miR-17-92 cluster gene edited by CRISPR/Cas9n (named as KTC2-Cl) were constructed according to the protocol of the laboratory of Ran et al. (21,22) by co-transfection of two plasmids containing guide RNAs (gRNAs) designed using the CRISPR Design website (
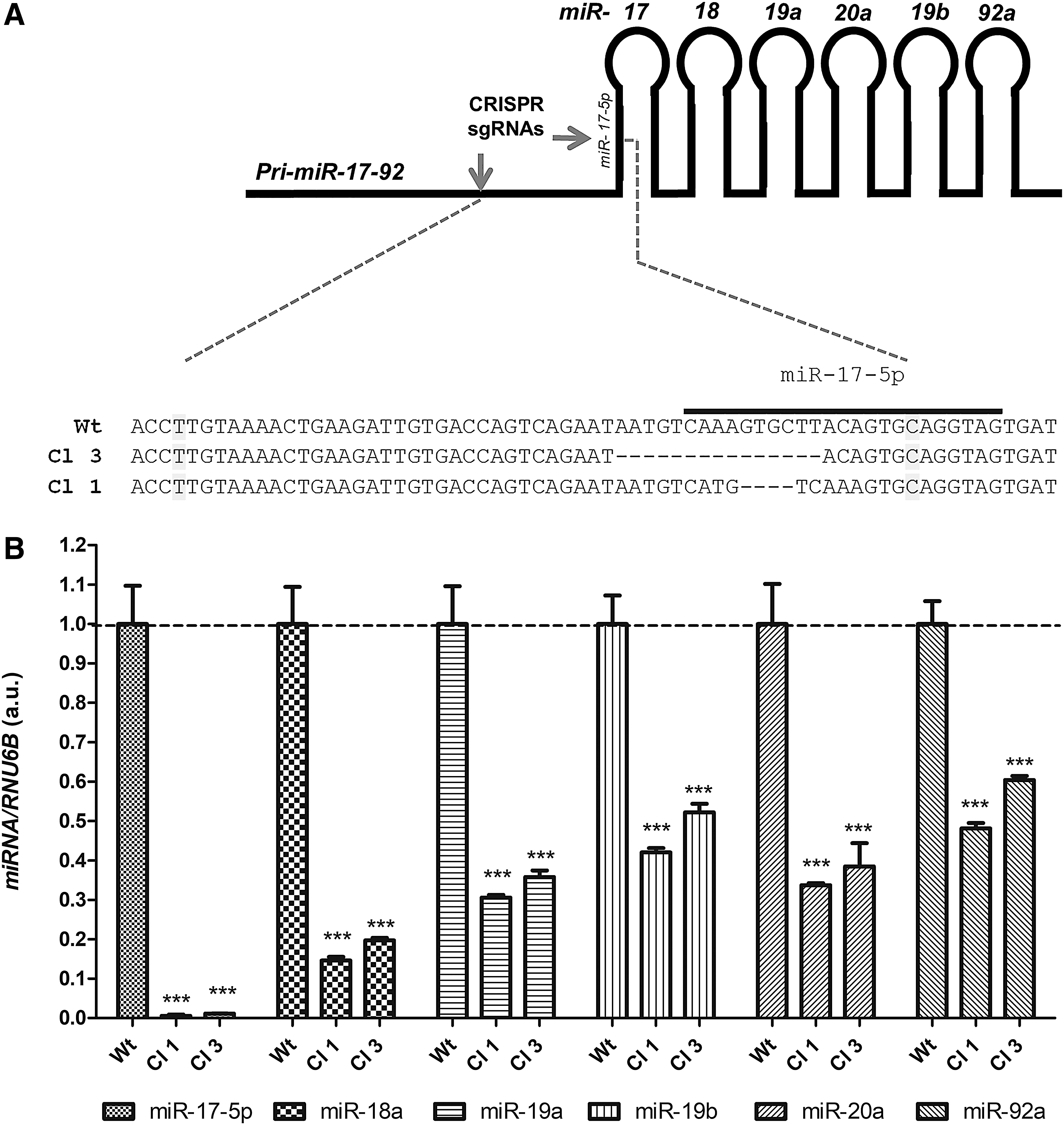
CRISPR/Cas9n-edited miR-17-92 gene in anaplastic thyroid cancer. (
Genomic DNA was used to amplify an 800 bp target segment surrounding the gRNAs target site in the miR-17-92 gene with specific primers (Surveyor primers) (Supplementary Table S1), and the Surveyor® Mutation Detection Kit (Integrated DNA Technologies, Skokie, IL) was used to confirm successful gene editing (mismatch) in the clones (Supplementary Fig. S1). GFP positive + puromycin-resistant single cell clones were expanded, and gDNA was extracted for sequencing confirmation after cloning the polymerase chain reaction (PCR) product into pGEM-T-easy (Promega, Madison, WI).
The rat PCCl3 thyroid follicular cell line was cultivated in F-12 Coon's modified medium (Sigma, St. Louis, MO) supplemented with 5% FBS and four hormones and maintained at 37°C and 5% carbon dioxide (CO2), as described previously (17). We transfected PCCl3 cells with different plasmids (Supplementary Table S2) to generate stable cell lines with expression of the whole cluster (PCCl3-miR-17-92), miR-17 (PCCl3-miR-17), miR-19b (PCCl3-miR-19b), miR-20a (PCCl3-miR-20a), miR-92 (PCCl3-miR-92a), and the segment with miR-19a/miR-20a/miR/19b (PCCl3-miR-19a/miR-20a/miR-19b). As control, we transfected PCCl3 cells with empty plasmid (PCCl3-Ø).
Cell function assays
Cell counting
Growth curves were determined using KTC2 cells by seeding 2 × 104 cells per well in 6-well plates and cultivation for 24–72 hours. TGF-β1 responsiveness was analyzed by seeding 5 × 104 KTC2 cells per well in 6-well plates. After 12 hours of adherence, cells received one pulse of recombinant TGF-β1 (rTGF-β1) (5 ng/mL) (rTGF-β1; Peprotech, Rocky Hill, NJ) in the culture medium. Cells were detached by trypsinization after 48 hours and counted in a Guava minicytometer (Millipore, Burlington, MA). High-concentration iodine treatment in PCCl3 cells was performed by diluting a 1 M stock solution of sodium iodide (NaI) in the medium to reach 10−3 M, and cell counting was performed after 24 hours of iodine treatment.
Cell viability assay (MTT)
Cells were plated at 2 × 104 cells per well in 96-well plates and cultured for 24 hours. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to the cell culture medium to a final concentration of 250 μg/mL and incubated at 37°C in 5% CO2 incubator for 4 hours. Then, the medium was removed and formazan crystals were solubilized with 0.01 M HCl in isopropanol. Absorbance was measured at 595 nm in a plate spectrophotometer SpectraMax M (Molecular Devices, San Jose, CA).
Cell cycle
Cells were detached by trypsinization, pelleted with supernatant (dead cells), and fixed with 70% ethanol. After hydration in phosphate-buffered saline (PBS), cells were treated with RNase (100 μg/mL) and DNA was stained with propidium iodide (50 μg/mL). The cell cycle was characterized by counting 10,000 events under flow cytometry using Guava EasyCyte Mini (Millipore).
Wound assay
1 × 105 cells were plated in 6-well plates and cultured until confluency. Next, 2 μg/mL mitomycin (Sigma) was added to the medium for 2 hours before performing the scratch in the monolayer using a 100-μL tip. After several washes with PBS, the medium was replaced by complete medium containing 2 μg/mL mitomycin (Sigma). Wound closure was monitored until cells completely repopulated the scratch (48 hours) under an inverted microscope EVOS XL Core Imaging System (Thermo Fisher Scientific, Waltham, MA).
Transwell assay
A migration assay was performed using uncoated Transwell™ chambers with membrane pore size of 8.0 μm (Corning Inc, Corning, NY). 2 × 104 cells were suspended in culture medium containing 0.5% FBS and plated in the upper chamber, whereas the lower chamber contained culture medium with 5% FBS. The cells that migrated through the membrane within 24 hours were fixed in 4% paraformaldehyde diluted in PBS and stained with 0.5% crystal violet. Cells were photographed using a Nikon Eclipse E600 microscope equipped with a CF160 epi-fluorescence optical camera and counted (10 representative fields).
Gene expression by quantitative PCR
Total RNA was extracted using the phenol–chloroform method using the TRIzol reagent (Invitrogen-Thermo Fisher, Carlsbad, CA). Protein coding gene expression was investigated using complementary DNA (cDNA) generated from the reverse transcription of 1 μg of total RNA using oligo-dT primer and MMLV reverse transcriptase (Invitrogen-Thermo Fisher). Quantitative PCR was performed in a ViiA7® Sequence Detection System (Applied Biosystems-Thermo Fisher, Foster City, CA) using SYBR Green Master Mix, cDNA, and specific primers (Supplementary Table S1). Gene expression was normalized by comparison with Rpl19 levels and calculated using the QGene program (23).
For miRNA expression, 10 ng of total RNA was reverse transcribed using a TaqMan® Reverse Transcription Kit (Applied Biosystems-Thermo Fisher) in the presence of stem-loop primers, followed by quantitative PCR using TaqMan MicroRNA Assays for miR-17-5p (assay 393), miR-18a-5p (assay 2422), miR-19a-3p (assay 395), miR-19b-3p (assay 396), miR-20a-5p (assay 580), miR-92a-3p (assay 430), small nucleolar RNA (snoRNA) (assay 1718), and RNU6B (assay 1093) (Applied Biosystems-Thermo Fisher), and TaqMan Universal PCR Master Mix, No AmpErase® UNG (Life Technologies-Thermo Fisher, Carlsbad, CA) in a ViiA7 Sequence Detection System. Gene expression was normalized by comparison to snoRNA levels and calculated using the QGene program (23).
Western blotting analyses
Total protein was extracted from cells using RIPA buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, and 0.1% SDS) containing 10% protease inhibitor cocktail (Sigma). Protein concentration was determined using the Bradford assay (Bio-Rad Laboratories, Hercules, CA), and 40 μg of each sample was fractionated by 10% SDS-PAGE and blotted onto a nitrocellulose Hybond-ECL membrane (Amersham Biosciences, Little Chalfont, UK). Nonspecific binding sites were blocked with 5% nonfat dry milk in Tris-buffered saline-0.1% Tween-20. The following primary antibodies were used: mouse anti-SMAD4 (sc-7866), rabbit anti-TGFBRII (sc-220), rabbit anti-TTF1 (sc-13040), mouse anti-PAX8 (sc-81353), rabbit anti-cMYC (sc-764), rabbit anti pSMAD3 (sc-130218), mouse anti-α-tubulin (sc-5286), mouse anti-β-actin (sc-47778), mouse anti-vimentin (sc-32322) (Santa Cruz, Santa Cruz, CA), and rabbit anti-LIN28B (Cell Signaling, Danvers, MA). The rabbit anti-NIS antibody was kindly donated by Dr. Sissy Jhiang. Antibody bound to target protein was detected with horseradish peroxidase-conjugated secondary antibodies and developed with luminol and p-coumaric acid (Sigma) reagents in the presence of hydrogen peroxide. Chemiluminescence emission was visualized with a ImageQuant LAS4000 imaging system (GE Healthcare, Little Chalfont, UK).
Bioinformatics and luciferase assay
miR-17 and miR-20 target validation
A segment of the rat Pax8 and human PAX8 3′-untranslated region (UTR) containing the predicted site miR-17/miR-20 (position 1079–1085 in rat and position 1990–1997 in human mRNA) extracted from TargetScan database (24) was cloned into pmiR-Glo plasmids (Promega) according to the manufacturer's instructions. Briefly, to generate the wild-type (Wt) miR-17/20 binding site reporter plasmid (pmiR-Glo-Pax8-Wt), two oligonucleotides were annealed and ligated in pmiR-Glo digested with XhoI and XbaI. As control, we also generated mutated plasmids that contained mutations in the seed sequence of miRNA binding sites (pmiR-Glo-Pax8-mut) that would block miRNA interaction. We also constructed a miR-19 and a miR-17/20 pmiR-Glo plasmid to test DICER1 as potential target (Supplementary Table S1). Transient transfection of pmiR-Glo-Pax8-Wt and pmiR-Glo-Pax8-mut was performed in PCCl-miR-17, PCCl3-miR-20a, and PCCl3-miR-19b cells. Forty-eight hours after transfection, cell lysates were collected and firefly luciferase activity was measured in a SpectraMax L Luminometer (Molecular Devices) using a Dual Luciferase® Reporter assay system (Promega). Luminescence was normalized by Renilla luciferase levels.
Nis, Tg, and Tpo promoter activity
For this luciferase reporter assay, we used the following plasmids: pNIS-2.8 (25) plasmid contains the 2854-bp fragment of the rat Nis promoter; pGL-Tg (26) contains a 202-bp fragment of the human TG promoter, and pTPO-luc contains a 420-bp fragment of the human TPO promoter (27). Three hundred nanograms of pNIS 2.8, pGL-Tg, or pTPO-luc plasmids was co-transfected with 30 ng of Renilla pRL plasmid as control for transfection efficiency using Lipofectamine 3000 (Life Technologies-Thermo Fisher). Cell lysates were collected after 24 hours of transfection. Firefly and Renilla luciferase were measured using the Dual-Luciferase Reporter assay system (Promega) in GloMax® 20/20 Luminometer (Promega).
TGF-β signaling activity
To evaluate the responsiveness to TGF-β signaling, we used the SBE4 Luc plasmid that contains four copies of the Smad Binding Element (SBE) or a p3TP-lux plasmid (Addgene) (Supplementary Table S2). Briefly, 4 × 104 cells were seeded into 24-well plates and transfected with 300 ng of SBE4-Luc (or p3TP-lux) plus 30 ng of pRL (Renilla luciferase) using Lipofectamine 2000 (Invitrogen-Thermo Fisher) before the treatment with 1 ng/mL (20 U/mL) rTGF-β (Peprotech) in culture medium for 24 hours. Cell lysates were collected after 24 hours, and luminescence was measured as described previously. To evaluate the effect of TGFBR2 knockdown on TGF-β signaling, we transiently transfected KTC2 cells with a SGEP plasmid containing short hairpin RNA for human TGFBR2 mRNA (Supplementary Table S1) 48 hours before transfection with TGF-β reporter plasmid SBE4 luc. Then, cells were treated with 1 ng/mL TGF-β1 for 24 hours, and cell lysates were collected and luminescence was measured as previously described.
Statistical analysis
The results are presented as the mean ± standard deviation, and they were submitted to analysis of variance followed by the Tukey test. Differences were considered significant at p < 0.05.
Results
miR-17-92 silencing via CRISPR/Cas9n induces thyroid follicular cell differentiation and restores TGF-β antimitogenic response
As miR-17-92 cluster expression is increased in ATC samples (12) and cell lines (Supplementary Fig. S2), we investigated the role of silencing miR-17-92 in thyroid cell function. To that end, we used the CRISPR/Cas9n system to efficiently edit the miR-17-92 gene in the human ATC cell line KTC2 and knock down all the cluster components simultaneously. Using two CRISPR/Cas9n plasmids, we designed two single guide RNAs to target the 5′ region upstream of the miR-17 precursor. This region contains the splicing factor binding site (Fig. 1A) essential for primary miR-17-92 processing into mature miRNAs (28). The edition of the miR-17-92 gene was efficient in ATC cell line KTC2 as observed in genomic DNA sequencing of two clones (KTC2-Cl1 and KTC2-Cl3) (Fig. 1A). The gene editing resulted in global downregulation of miR-17-92 mature miRNA levels, ranging from >90% inhibition of miR-17-5p to >40% inhibition for miR-92a (Fig. 1B).
After this initial validation of the system, we evaluated expression of thyroid differentiation genes in response to miR-17-92 silencing by CRISPR/Cas9n. We observed an increase in the gene expression of NIS, TPO, TG, and the transcription factors PAX8 and NKX2-1 in KTC2-Cl1 and KTC2-Cl3 (Fig. 2A). NIS mRNA expression was detected at very low levels that increased after miR-17-92 silencing. Accordingly, protein expression of PAX8 and NKX2-1 was increased in KTC2-Cl1 and KTC2-Cl3 analyzed by Western blot (WB) (Fig. 2B). However, NIS and TPO protein were undetectable by WB and immunofluorescence (data not shown).
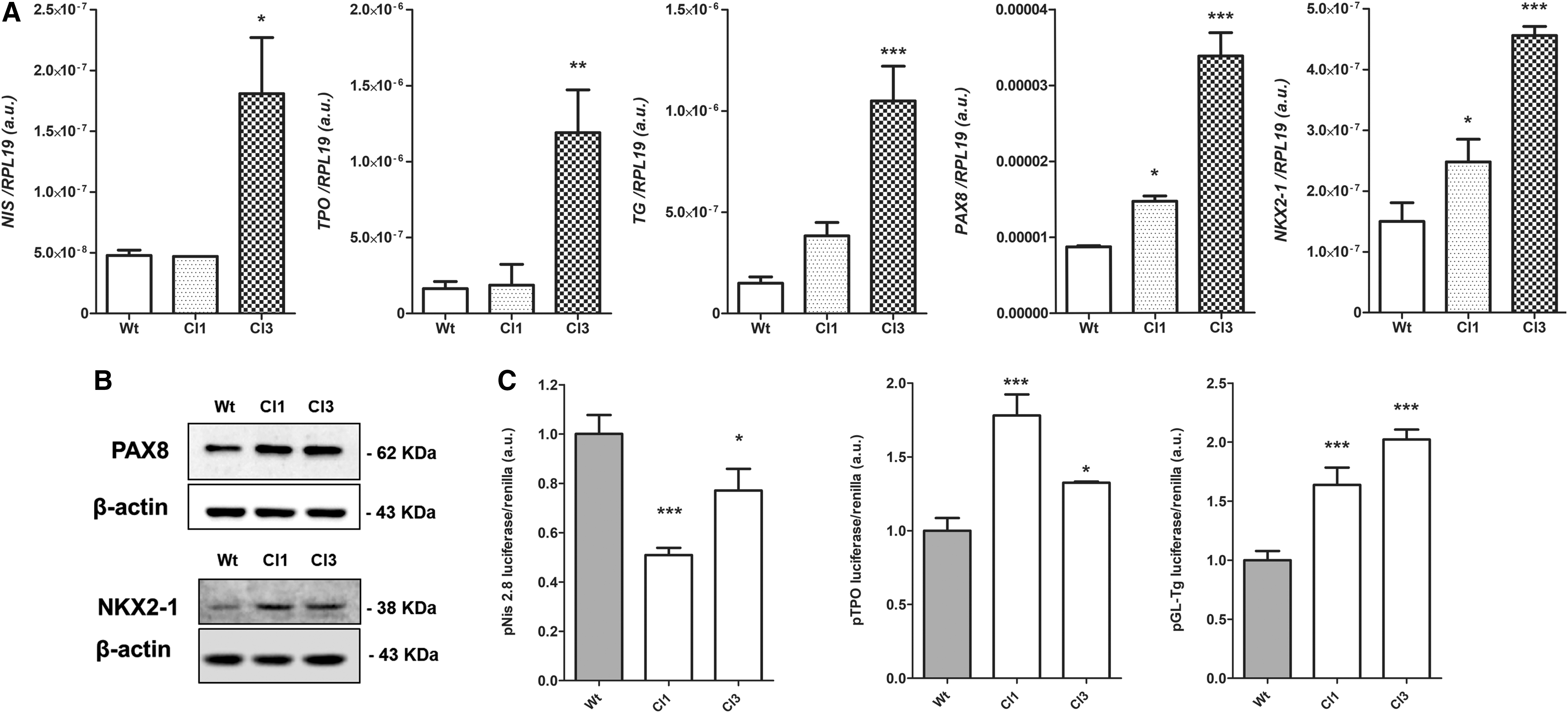
Effect of CRISPR/Cas9n editing of mir-17 gene in thyroid cell differentiation. (
Next, we used the luciferase reporter plasmids pNis 2.8, pGL-Tg, and Tpo-luc to investigate promoter activity of the NIS (SLC5A5), TG, and TPO genes, respectively. We observed a strong induction of TG and TPO promoters in miR-17-92 CRISPR/Cas9n-edited KTC2-Cl1 and KTC2-Cl3, while interestingly NIS gene promoter activity was reduced compared with KTC2-Wt cells (Fig. 2C).
Functionally, targeting miR-17-92 by CRISPR/Cas9n exerted an antitumorigenic effect in KTC2 cells. We observed a reduced cell count of KTC2-Cl1 and KTC2-Cl3 clones compared with KTC2-Wt cells (Fig. 3A). Moreover, cell colony formation (Fig. 3B) and cell migration (in the presence of mitomycin) (Fig. 3C) were strongly repressed in KTC2-Cl1 and KTC2-Cl3 clones, leading us to investigate TGF-β signaling, an important antiproliferative pathway for thyroid cells. We observed increased protein levels of the TGF-β signaling components TGFBR2 and SMAD4 (Fig. 3D), both validated targets of miR-17-92 (17,29), in KTC2-Cl1 and KTC2-Cl3 clones compared with KTC2-Wt cells. Interestingly, MYC protein levels were reduced in KTC2-Cl1 and KTC2-Cl3 clones (Fig. 3D). MYC is a known positive regulator of miR-17-92 (30) and is repressed by TGF-β signaling activation (31).
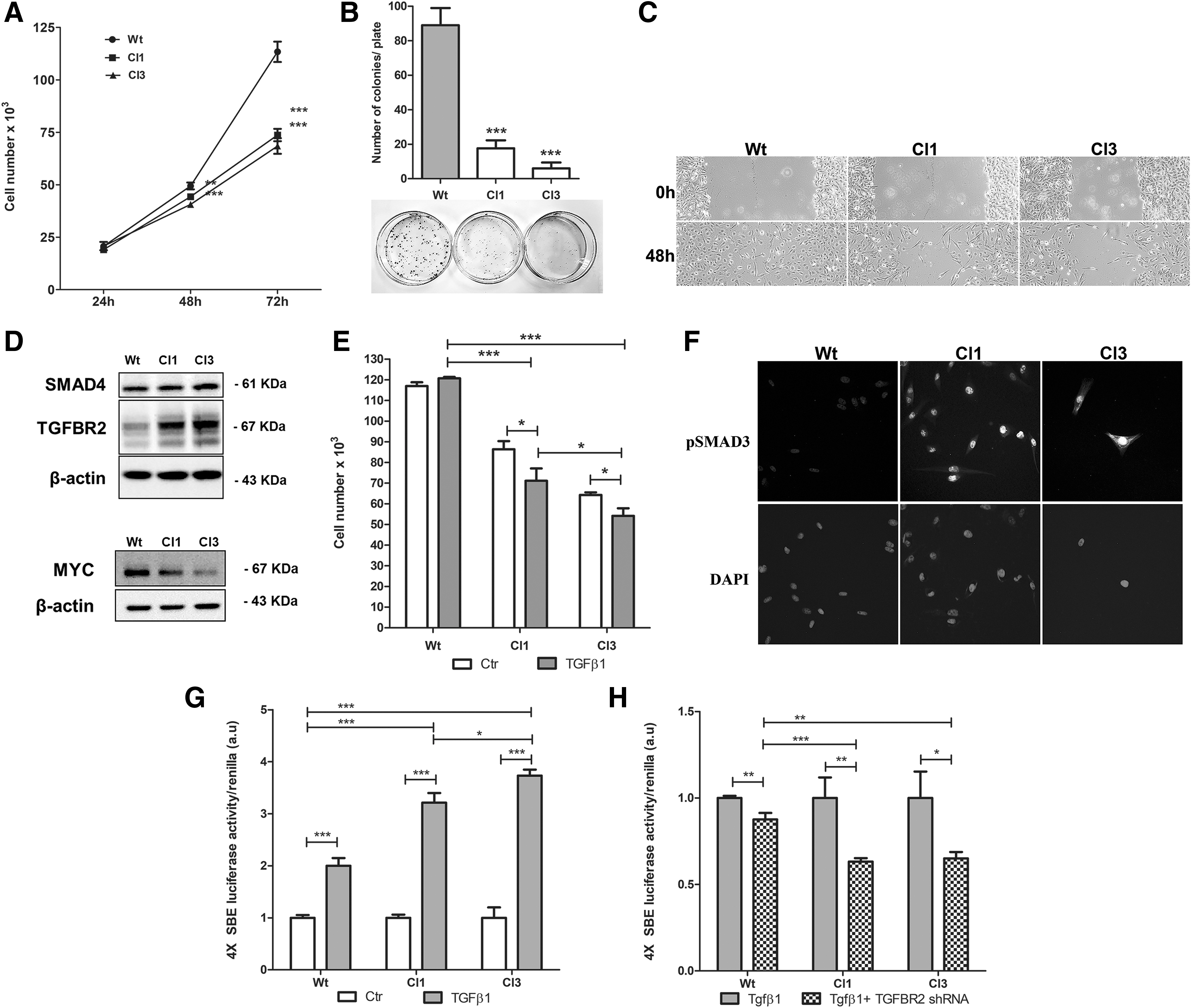
Effect of miR-17-92 gene editing in KTC2 thyroid cell biology. (
To evaluate whether TGF-β signaling responsiveness was altered in KTC2 clones after miR-17-92 edition, we performed cell counting in the presence of recombinant active TGF-β1 and observed that KTC2-Cl1 and KTC2-Cl3 showed recovery of the antimitogenic response to TGF-β1 treatment leading to reduction in cell number, an effect not observed in KTC2-Wt cells (Fig. 3E). Indeed, KTC2-Cl1 and KTC2-Cl3 showed high basal levels of pSMAD3 nuclear staining compared with KTC2-Wt indicating a reactivation of TGF-β signaling in these cells (Fig. 3F). This finding was further confirmed by a SBE4 luciferase reporter assay that showed a 60% and 80% higher response after recombinant TGF-β1 treatment in KTC2-Cl1 and KTC2-Cl3, respectively, compared with KTC2-Wt cells (Fig. 3G).
miR-17-92 overexpression impairs TGF-β signaling in normal thyroid follicular cells
To investigate the role of miR-17-92 in normal thyroid cell biology, we overexpressed the miR-17-92 cluster in normal rat thyroid follicular cells PCCl3 (stable cell line PCCl3-miR-17-92) (Fig. 4A). miR-17-92 overexpression increased cell viability measured by MTT (Fig. 4B) and resulted in a fourfold increase in cell migration in PCCl3-miR-17-92 cells evaluated by a transwell assay (Fig. 4C), although the cell count was not significantly altered (data not shown).
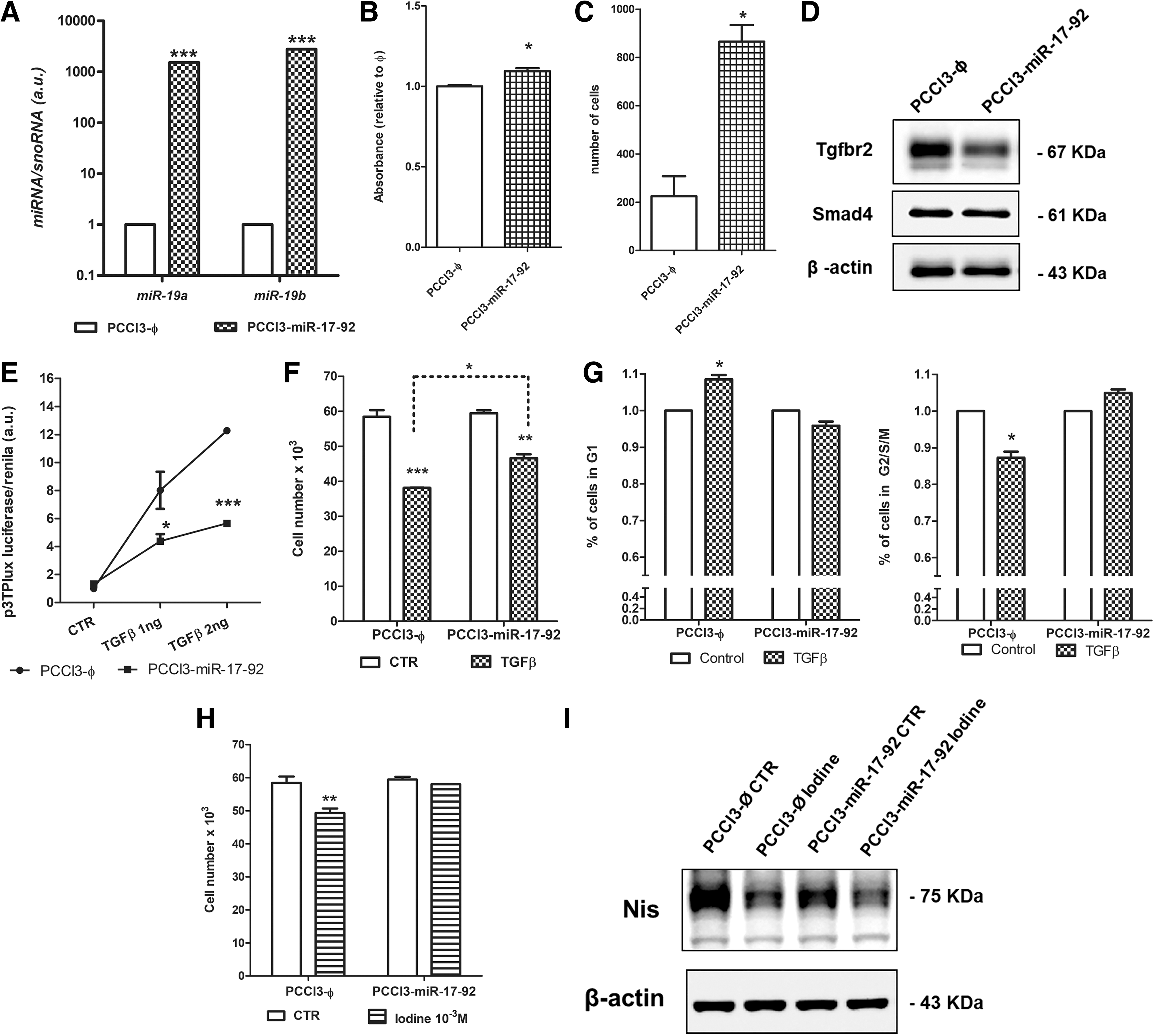
Effects of miR-17-92 overexpression in normal rat thyroid follicular cells PCCl3. (
As miR-17-92 targets are enriched in the TGF-β signaling pathway, we analyzed the protein levels of TGFBR2 and SMAD4 and observed a reduction of TGFBR2 in PCCl3-miR-17-92 (Fig. 4D). To investigate whether TGFBR2 reduction would impact TGF-β signaling, we used a 3TP-lux luciferase reporter assay to evaluate TGF-β responsiveness. We observed that the treatment of control PCCl3-Ø cells with recombinant active TGF-β1 resulted in a dose-dependent increment in luciferase reporter activity (Fig. 4E). However, this effect was significantly reduced by miR-17-92 overexpression in PCCl3-miR-17-92 cells (Fig. 4F). Moreover, TGF-β1 treatment resulted in a smaller reduction in cell counting for PCCl3-miR-17-92 cells compared with control PCCl3-Ø (Fig. 4F). The analysis of the cell cycle showed that TGF-β1 treatment in PCCl3-miR-17-92 cells does not induce G1-cell cycle arrest as observed in PCCl3-Ø cells (Fig. 4G).
To further evaluate the role of miR-17-92 in TGF-β signaling, we next tested the effect of a high iodine concentration on the cell count of PCCl3-miR-17-92 cells. High iodine induces an antiproliferative effect that is dependent on the activation of the TGF-β pathway (32). We observed that high iodine concentration (10−3 M NaI) decreased the cell count in control cells (PCCl3-ϕ), but this effect was attenuated after overexpression of miR-17-92 in PCCl3-miR-17-92 cells (Fig. 4H). Next, we investigated the influence of NIS in this effect as this protein is essential to iodine trapping in thyroid follicular cells. Surprisingly, Western blotting showed that PCCl3-miR-17-92 cell exhibited reduced NIS levels (Fig. 4I) that could contribute to the refractoriness to high iodine treatment. Altogether, these results show that miR-17-92 activation in normal thyroid follicular cells exerts a pro-proliferative effect by blunting TGF-β signaling responsiveness.
miR-17-92 overexpression impairs thyroid follicular cell differentiation in normal thyroid follicular cells
The expression analysis of genes involved in thyroid differentiation showed that overexpression of miR-17-92 severely repressed Nis and Tpo gene expression, which are related to iodine metabolism, as well as the thyroid transcription factors Nkx2-1 and Pax8 in PCCl3-miR-17-92 cells (Fig. 5A). The analysis of Nis (Slc5a5) and Tg promoter activity using a luciferase reporter assay showed that the Nis and Tg genes are transcriptionally repressed after miR-17-92 overexpression in PCCl3 cells (Fig. 5B). At the protein level, we also observed an important reduction in NIS and PAX8, while no modulation was observed in NKX2-1. Moreover, miR-17-92 induced the expression of vimentin and LIN28B, markers for mesenchymal cells and stem cells, respectively (Fig. 5C).
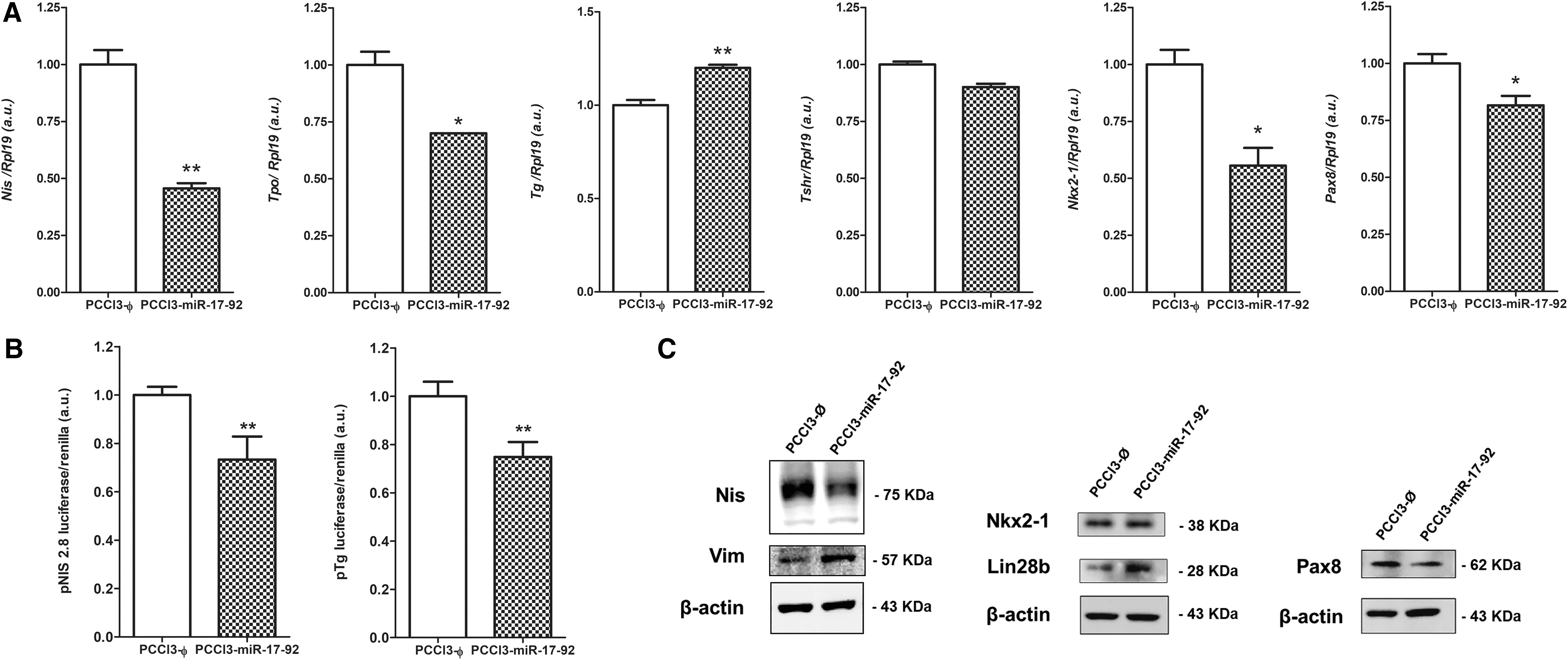
Overexpression of miR-17-92 cluster reduces differentiation genes in normal rat thyroid follicular cells. (
miR-17-92 cluster components modulate thyroid differentiation
To dissociate the role of individual miRNAs that comprises the miR-17-92 cluster, we transfected PCCl3 cells with plasmids to stably express miR-17, miR-19b, miR-20a, miR-92a individually and the miR-19a/miR-20a/miR-19b cluster as a group (Fig. 6A). We observed that individually miR-19b, miR-92a, and miR-17 strongly inhibited the expression of NIS, while miR-20a showed no effect on NIS protein expression (Fig. 6B). Indeed, the combined effect of the whole miR-17-92 cluster components in PCCl3-miR-17-92 resulted in NIS downregulation (Fig. 5C). For the thyroid transcription factor PAX8, we observed that individually miR-17, miR-19b, and miR-20a inhibited PAX8 protein levels (Fig. 6B) resulting in PAX8 inhibition when the whole miR-17-92 cluster is overexpressed (Fig. 5C) in PCCl3 cells. However, the expression of individual or combined miR-17-92 components did not change NKX2-1 protein levels (Figs. 5C and 6B).

Overexpression of miR-17-92 cluster components reduces differentiation genes in normal rat thyroid follicular cells. (
In this context, deregulation of thyroid transcription factor levels could impair NIS protein by influencing the transcriptional activity of the Nis gene. To investigate this possibility, we used the reporter plasmid pNis 2.8 as it contains a 2.8-Kb fragment of the rat Nis gene promoter comprising two PAX8 and one NKX2-1 site (25). Indeed, the activation of the miR-17-92 cluster components lead to transcriptional impairment of the Nis gene and a reduction in NIS levels in normal thyroid cells PCCl3 (Fig. 6C). miR-19 expression per se or in combination (miR-19a/miR-20a/miR-19b), and miR-17 exerted important repressive effects on Nis gene transcription. For miR-20a, we did not observe any significant modulation of Nis gene transcription, a result that was confirmed by analyzing NIS expression by WB. Interestingly, miR-92a slightly induced Nis transcriptional activity (Fig. 6C). We observed similar results when analyzing Tg promoter activity using a pGL-Tg reporter plasmid in PCCl3 cells, showing transcriptional inhibition when miR-17, miR-19 per se, or in combination (miR-19a/miR-20a/miR-19b) were overexpressed (Fig. 6C). Overall, the expression of the whole miR-17-92 cluster components leads to transcriptional inhibition of the Nis and Tg promoters in normal thyroid follicular cells PCCl3 (Fig. 5B).
These results indicate that miR-17-92 exerts its effects on thyroid differentiation through a common mechanism mediated by thyroid transcription factors. Consequently, we used an in silico approach to search for miR-17-92 cluster targets relevant to thyroid cell function. Indeed, analysis with the TargetScan tool revealed that the 3′-UTR segment of rat Pax8 mRNA contains predicted sites for miR-17-5p and miR-20a-5p. To test the interaction of these miRNAs and Pax8 3′-UTR, we constructed a reporter plasmid that contained the Wt sequence of the rat Pax8 3′-UTR (position 1079–1085 in the 3′-UTR sequence) predicted to interact with miR-17-5p and miR-20a-5p, cloned downstream of Luciferase gene in pmiR-Glo plasmid. We also constructed a mutated plasmid (mut) containing mismatches in the region of miRNA/mRNA pairing as control. We transfected these plasmids into PCCl3 cells overexpressing single components of the cluster, miR-17, miR-20a, and miR-19b (specificity control) and did not observe a significant downregulation of luciferase activity neither in miR-17-expressing cells nor in miR-20a-expressing cells (data not shown). This result indicates that in this model Pax8 mRNA is not a target for miR-17-5p/miR-20a-5p.
Moreover, studies using different mouse models for Dicer1-specific knockout in thyroid follicular cells have shown that miRNAs are necessary to maintain thyroid cell differentiation and function. In these models, loss of DICER1 leads to severe hypothyroidism, and a loss of thyroid cell differentiation and proliferation (33,34). The in silico search also showed that rat Dicer1 mRNA 3′-UTR contains one predicted site for an interaction with miR-17-5p/miR-20-5p and another conserved site for miR-19-3p. We also constructed a pmiR-Glo plasmid containing the interaction site for miR-17-5p/miR-20a-5p (position 2722–2728) and miR-19-3p (position 1324–1330). However, our results did not show any significant modulation of these Dicer1 3′-UTR luciferase reporters in response to the modulation of miR-17-5p/miR-20a-5p and miR-19-3p (data not shown), indicating that in this model, Dicer1 mRNA is not a target for these miRNAs.
Discussion
Overexpression of miR-17-92 is detected in several types of cancers as an important marker of malignancy and tumor aggressiveness (13,35). Indeed, high levels of miR-17-92 are detected in aggressive ATC (12). We show in this study that CRISPR/Cas9n-mediated editing of the miR-17-92 gene is efficient and results in a strong inhibition of miR-17-92 mature miRNAs in the ATC cell line. We designed the gRNAs to target the upstream region of miR-17 pre-miR that contains a splicing site essential for pri-mir-17-92 processing (28). Importantly, we observed a recovery of the expression of the thyroid-specific genes TPO and TG, and to some extent NIS, as well as the PAX8 and NKX2-1 transcription factors. Reestablishing NIS expression is one of the biggest challenges of treating aggressive thyroid cancers. NIS suppression in thyroid cancer is multifactorial and related to epigenetics, transcriptional, and post-transcriptional regulation that involves DNA methylation, MAPK signaling, and miRNAs (18,36,37).
The loss of expression of thyroid differentiation markers related to iodine metabolism is a key event during thyroid oncogenesis and is correlated with aggressive tumor behavior, such as radioiodine refractoriness (RAIR). Ultimately, RAIR is caused by the loss of the iodine-trapping ability mediated by NIS downregulation. Indeed, RAIR thyroid tumors display a high frequency of BRAF mutation and alterations in the RAS and PI3K signaling pathways (19). Activation of the BRAFV600E oncogene is a well-known inhibitor of NIS and thyroid cell differentiation (18). Our group showed recently that the BRAFV600E mutation increases miR-17-92 expression in normal thyroid follicular cells and reduces TGF-β signaling responsiveness (17). Moreover, the BRAFV600E oncogene can be associated with aggressive clinical–pathological characteristics, such as extrathyroidal invasion, and mortality in thyroid cancers (38 –40).
In our study, we show that the activation of the miR-17-92 cluster is an important step to downregulate NIS and thyroid cell differentiation, leading to a loss of iodide uptake in vitro and, hence, expected to be associated with refractoriness to radioiodine in vivo. We dissected the role of individual miRNA components of the miR-17-92 cluster in thyroid differentiation, especially the impact on NIS levels, and observed that while some miRNAs such as miR-17-5p, miR-19a/b, and miR-92 may inhibit NIS expression, others such as miR-20a-5p do not modulate NIS levels. Moreover, miR-17, miR-19, and miR-20a downregulate the PAX8 transcription factor, indicating that a cooperative targeting of thyroid differentiation genes and transcription factors by the miR-17-92 cluster is a key effect of this oncomiR in thyroid cancer. Taken together, the single components of the miR-17-92 cluster have inhibitory and stimulatory effects in thyroid differentiation, but the overexpression of the whole miR-17-92 cluster results in an overall inhibitory effect in thyroid follicular cells.
Recently, other miRNAs have been implicated in the regulation of NIS and thyroid differentiation such as miR-339-5p and miR-146b-3p (41,42). miR-339-5p is increased in human PTC and targets the 3′-UTR of NIS mRNA, and its overexpression efficiently reduces NIS mRNA levels and radioiodine trapping in PCCl3 and MCF7 cells (41). Moreover, miR-146b-5p is the most overexpressed miRNA in PTC and a hallmark of thyroid cancer, and the 3p strand (miR-146b-3p) mediates the targeting of NIS and PAX8 mRNAs, leading to the downregulation of thyroid differentiation (42).
In this study, we also show that miR-17-92 activation leads to a loss of responsiveness to the antimitogenic effect of TGF-β in normal thyroid cells via deregulation of TGF-β signaling components, such as TGFBR2 and SMAD4. In a previous study, we showed that BRAFV600E oncogene induces miR-19a/b that targets SMAD4 mRNA (17), an effect also observed in SMAD4 when we overexpressed miR-19b in PCCl3 cells (data not shown).
Moreover, silencing of the mature miR-17-92 in KTC2 cells using CRISPR/Cas9n reduced cell counts and increased SMAD4 and TGFBR2 levels, known targets of miR-17 and miR-19 (29), which led to partial restoration of the antimitogenic effect upon rTGF-β1 treatment. Interestingly, cMYC protein levels were reduced after miR-17-92 gene silencing. cMYC is one of the transcriptional regulators of miR-17-92 (30), and studies have shown that miR-17-92 cooperates with/mediates MYC effects to promote tumorigenesis (14,43). However, reduction of MYC levels could be associated with the reactivation of TGF-β signaling, which has been shown to repress cMYC transcription (31).
In this study, we found a pro-oncogenic role of the miR-17-92 cluster associated with a loss of differentiation, a loss of NIS expression, and refractoriness to the antiproliferative signal of TGF-β. By dissecting the cluster into separate miRNAs, we demonstrate that miR-19, miR-92, and miR-17 exert important repressive effects on NIS, NKX2-1, and PAX8 protein levels. Targeting miR-17-92 via CRISPR/Cas9n gene editing resulted in an effective and stable reduction of mature miR-17-92 and induced a partial restoration of a differentiated phenotype characterized mainly by reestablishing expression of NIS, TG, TPO, PAX8, and NKX2-1. Moreover, it restored TGF-β signaling responsiveness in an ATC cell line. The success of CRISPR/Cas9 editing to correct diseases in animal models and its recent success in human embryos (44 –46) may ultimately open new perspectives for patients with incurable or aggressive diseases, such as cancer.
Conclusions
miR-17-92 is a potent regulator of thyroid follicular cell differentiation, and CRISPR/Cas9n-mediated gene editing is efficient to silence miR-17-92 expression in an ATC cell line, resulting in improvement of thyroid differentiation and TGF-β antiproliferative response while inducing antitumorigenic effects. Thus, molecular targeting of miR-17-92 could be a potential therapeutic approach for aggressive thyroid carcinomas.
Footnotes
Acknowledgments
The authors would like to thank the kind donation of KTC2 cells by Dr. Norisato Mitsutake (Nagasaki University Graduate School of Biomedical Sciences, Nagasaki, Japan); donation of the plasmids pNIS 2.8 and pGL-Tg by Dr Pilar Santisteban (Instituto de Investigaciones Biomédicas, CSIC-UAM, Madrid, Spain) and pTPO-luc by Dr. Samuel Refetoff (The University of Chicago, IL); and donation of NIS antibody by Dr. Sissy M. Jhiang (The Ohio State University, Columbus, OH).
Author Disclosure Statement
The authors declare that there is no conflict of interest that would impair the impartiality of this article.
Funding Information
This study was supported by scholarships and grants from Sao Paulo Research Foundation (FAPESP) numbers 2014/50521-0, 2016/17129-4, 2017/01829-0, the National Council for Scientific and Technological Development (CNPq) numbers 308331/2017-6, 151789/2018-5, 430756/2018-6, and University of Sao Paulo, NAPmiR Research Support Center.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2