
Abstract
Background:
Thyroid cancer patients with radioiodine-refractory (RAI-R) disease, resulting from insufficient RAI delivery and/or RAI resistance, have increased mortality and limited treatment options. To date, studies have largely focused on tumor mutations associated with different stages of disease, which could provide prognostic value for RAI-R disease. It was hypothesized that germline variants contributing to intrinsic differences in iodine metabolism, tumor microenvironment, and/or immune surveillance are associated with RAI-R disease.
Methods:
Whole-genome genotyping data analysis was performed on 1145 Caucasian (CAU) patients, 244 of whom were RAI-R, and 55 African American (AA) patients, nine of whom were RAI-R. Germline-variant association studies were conducted using candidate genes involved in iodine metabolism or DNA-damage repair, as well as genome-wide association analysis. Initial data indicated several notable variants in a small number of patients (n = 7), who were later determined to be AA patients of >80% African ancestry (n = 37). This led to the study focusing on germline single nucleotide polymorphisms uniquely associated with RAI-R AA patients. Sanger sequencing was performed to validate risk alleles and identify the incidence of the common somatic mutations BRAFV600E , NRASQ61R , and HRASQ61R in AA patients whose primary tumor samples were available (28/55).
Results: TG
, BRCA1, and NSMCE2 haplotypes were identified as being uniquely associated with RAI-R AA patients of >80% African ancestry. All patients with the TG haplotype (n = 4) had a biochemical incomplete response to RAI therapy. Patients with the NSMCE2 haplotype (n = 4) were diagnosed at a young age (13, 17, 17, and 26 years old) with distant metastatic disease at initial diagnosis. The BRCA1 haplotype co-occurred in three out of four patients with the NSMCE2 haplotype. The incidence of BRAFV600E appears lower in papillary thyroid carcinomas from AA patients of >80% African ancestry (3/14; 21%) than in AA patients of <80% African ancestry (6/9; 67%), albeit only just approaching statistical significance (p = 0.077). The tumors available from three RAI-R AA patients were negative for BRAFV600E , NRASQ61R , and HRASQ61R.
Conclusions:
The identification of candidate RAI-R risk haplotypes may allow early stratification of clinical manifestations of RAI-R disease followed by early intervention and personalized treatment strategies. Functional annotation of candidate RAI-R risk haplotypes may provide insights into the mechanisms underlying RAI-R disease.
Introduction
Thyroid cancer is the most common endocrine-related cancer, with an increasing global incidence rate (1). It is estimated that there were 805,750 people living with thyroid cancer in the United States in 2016, and an additional ∼60,000 new patients will be diagnosed yearly. The ability of thyroid follicular cells to concentrate and organify iodide allows the use of radioactive iodide (RAI) for imaging and targeted killing of RAI-avid residual and metastatic thyroid cancer after thyroidectomy. The majority of patients with differentiated thyroid cancer do well after standard therapy (surgery, 131I, thyrotropin suppression by levothyroxine replacement). However, 5–15% of patients become RAI refractory (RAI-R) and no longer benefit from 131I therapy (2,3). No novel treatment has shown improved overall survival for patients with progressive RAI-R disease, despite improved progression-free survival in some patients treated with multitargeted kinase inhibitors (4 –7).
Clinical manifestations of RAI-R disease appear as a spectrum of insufficient RAI uptake and/or RAI resistance (8,9). Genetics underlying the loss of RAI uptake in thyroid cancer have been extensively studied (10). MAPK activation was identified as an actionable target, and both BRAF and MEK inhibitors have been shown to restore or enhance the RAI uptake of metastatic lesions in some patients with non-RAI-avid disease (5,7). The genetics underlying RAI resistance in thyroid cancer has not been investigated, as the correlation between RAI uptake and responsiveness of lesions to 131I therapy has not been quantitatively evaluated. BRAFV600E accounts for approximately 60% of all driver mutations in papillary thyroid carcinomas (PTC) and is associated with an increased incidence of RAI-R disease (11). However, the identification of a tumor driver mutation alone is insufficient to predict progression to RAI-R disease for individual patients.
It is well recognized that the tumor microenvironment (12) and immune surveillance, which cannot be predicted based on specific tumor mutations, play essential roles in tumor progression and therapeutic responsiveness. It was hypothesized that germline variants giving rise to intrinsic differences in iodine metabolism, DNA-damage repair, and/or immune response may predispose patients to the acquisition of phenotypes conferring insufficient RAI uptake and/or RAI resistance contributing to RAI-R disease. This study aimed to identify germline variants associated with RAI-R disease, which may provide additional information, together with tumor driver mutations, to identify patients at risk of RAI-R disease and/or to stratify the diverse clinical manifestations of RAI-R disease.
Methods
Subjects
All studies were approved by the Institutional Review Board (IRB) at the Ohio State University (OSU), MD Anderson Cancer Center (MDACC), and the National Cancer Institute (NCI). Patients in the OSU cohort included in this study consented to participate in the Endocrine Neoplasia Repository (ENR), with patient accrual beginning in August 2006 to the present (n = 3139). In 2016, germline genotyping by deCODE Genetics (Iceland) (13) was performed on DNA from 1714 patients with non-medullary thyroid cancer. Of these, 1351 patients received RAI therapy, with sufficient clinical information for RAI-R categorization available for 1263 patients. The OSU cohort comprised patients with the following histologies: classic PTC (n = 680), follicular variant PTC (n = 221), other PTC variants (n = 177), follicular thyroid carcinoma (FTC; n = 176), poorly differentiated thyroid carcinoma (PDTC; n = 2), anaplastic thyroid carcinoma (ATC; n = 3), and unknown histology (n = 4). To validate the preliminary findings, blood DNA samples were obtained from 131I-treated, self-reported African American (AA) thyroid cancer patients who previously consented to be part of existing biorepositories at MDACC and the NCI. These cohorts included RAI-R and non-refractory (NON-R) patients from MDACC (n RAI-R = 5, n NON-R = 9) and NCI (n RAI-R = 1, n NON-R = 6). Unless explicitly stated to the contrary, all of the performed analyses and results refer specifically to the OSU cohort.
Genotyping and informatics
Whole-genome genotyping of blood DNA samples from 1351 thyroid cancer patients who had received RAI therapy was performed by deCODE Genetics (Iceland) (13) using the Omni-1 Quad-bead chip arrays (Illumina). Imputation was performed using 1000 Genomes Phase 3 data. Haplotype analysis with candidate single nucleotide polymorphisms (SNPs) for the OSU cohort was performed. Frequencies of the risk haplotypes in the 1000 Genome African population were estimated by downloading phased genotypes with DataSlicer (
Inference of genetic ancestry
Genetic ancestry was estimated for all the samples of the OSU cohort by applying the ADMIXTURE v1.30 (15) program, adopting a maximum likelihood algorithm, modeling the probability of observed genotypes using ancestry proportions and population allele frequencies to approximate both the allele frequency in a given population and the ancestry proportion of each sample simultaneously. SNP genotypes from Omni-1 Quad-bead Illumina chip data, after pruning with PLINK (16) to eliminate any LD structure with an r 2 cutoff of 0.5 in each sliding window of 50 SNPs and advanced by five SNPs, were selected. These SNPs were further filtered to remove any violation of the Hardy Weinberg Equilibrium (HWE) at 1.0 × 10–6. Genotypes of the remaining SNPs were used in the ADMIXTURE program with three clusters (k = 3) to estimate ancestry proportions for each individual.
Categorization of RAI-R disease
Patients were categorized as either RAI-R, with high or moderate confidence, or NON-R. Patients with insufficient clinical information to categorize their RAI-R disease status were excluded from the study (88/1351). AA and Caucasian (CAU) patients were categorized as RAI-R with high confidence (n AA = 6/55, n CAU = 112/1145) if they (i) received a cumulative 131I activity of ≥600 mCi, (ii) had continuously increasing serum thyroglobulin (Tg) levels in follow-up tests conducted more than six months after 131I treatment, or (iii) received additional surgical intervention and 131I treatment(s) following the initial thyroidectomy and 131I treatment, along with one or more of the moderate confidence criteria described below. Patients were categorized as RAI-R with moderate confidence (n AA = 3/55, n CAU = 132/1145) if they (i) received two or more consecutive 131I treatments of ≥100 mCi each that were more than six months apart, (ii) had unstimulated serum Tg >2 ng/mL or stimulated serum Tg >10 ng/mL persisting more than six months after 131I treatment, and/or (iii) received additional surgical intervention, such as lymphadenectomy, following initial thyroidectomy and 131I treatment.
Association analyses and candidate SNP selection
Patients with insufficient clinical information for RAI-R categorization (88/1351), CAU patients of <85% CAU ancestry (63/1351), and AA patients of <25% African ancestry (0/1351) were excluded from the candidate gene association analysis to reduce the potential of population stratification bias. However, no ancestry thresholds were applied to the GWAS to increase the total number to contend with the increased statistical stringency of multiple testing correction. The workflow for the association analyses is briefly summarized in Figure 1. The candidate gene association analyses were performed using selected DNA-damage repair (n = 86) and iodine metabolism (n = 15) genes. As RAI-R disease is phenotypically heterogeneous, four separate association analyses of differential RAI-R categorization confidence were performed to identify candidate variant reproducibility.
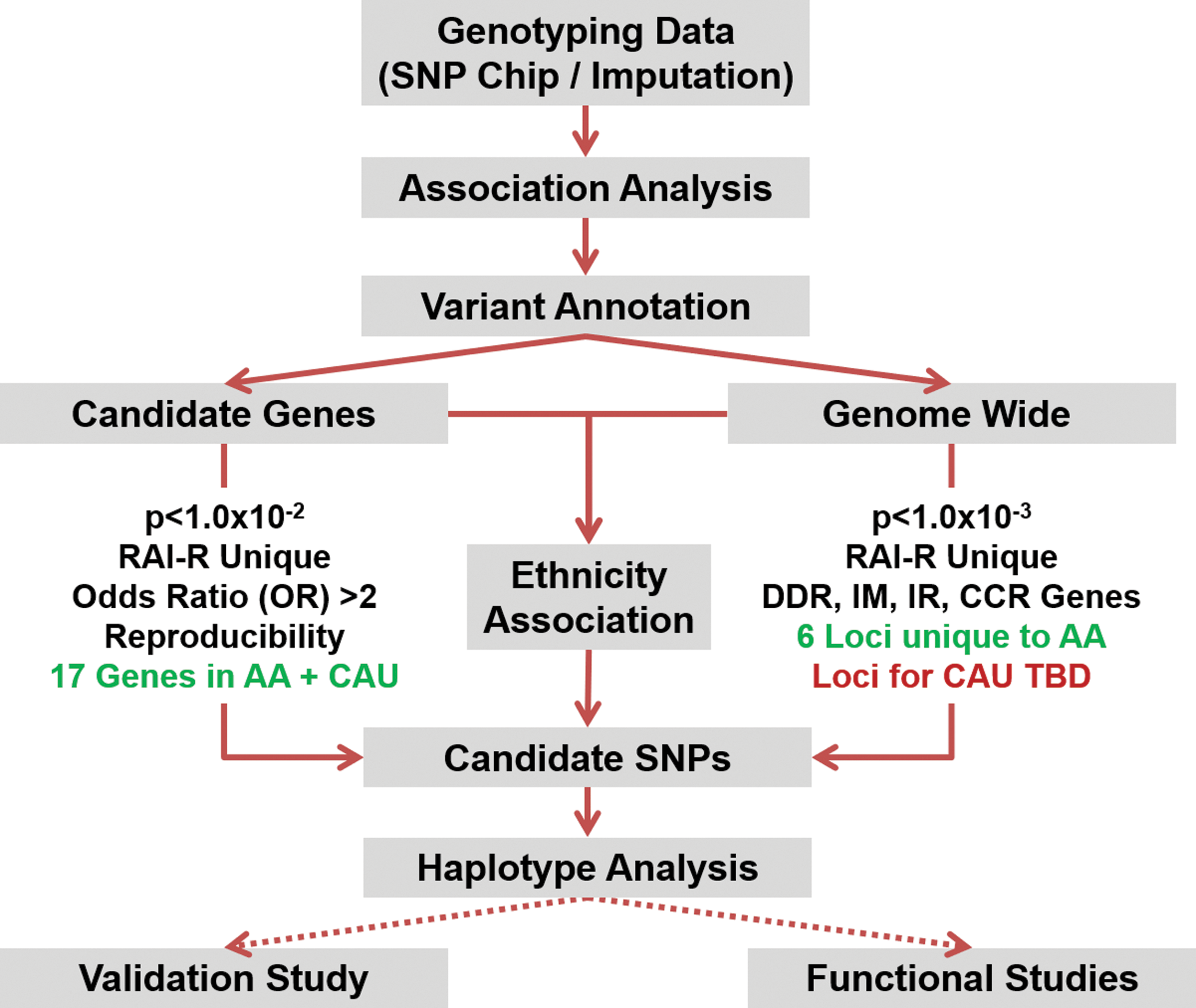
Work flow for candidate gene and genome-wide association analyses. Association analysis was performed on genotyping data derived from the Omni-1 Quad-bead single nucleotide polymorphism ChIP array or imputation. Subsequently, candidate variants were functionally annotated using ANNOVAR, and variant effects were predicted using SIFT, PolyPhen, and Mutation Taster. Candidate variants were selected based on statistical significance and unique association with radioiodine-refractory (RAI-R) patients. ADMIXTURE analysis was conducted to determine the association between candidate RAI-R risk variants and patient ethnicity. Future validation and functional studies of candidate variants remain to be conducted (dotted lines). AA, African American; CAU, Caucasian; DDR, DNA-damage repair; IM, iodine metabolism; CCR, cell-cycle regulation; TBD, to be determined.
The candidate gene association analyses included: (i) high confidence RAI-R CAU patients versus NON-R CAU patients (n RAI-R,CAU = 112 vs. n NON-R,CAU = 901), (ii) high confidence RAI-R CAU and AA patients versus NON-R CAU and AA patients (n RAI-R,CAU + AA = 118 vs. n NON-R,CAU + AA = 947), (iii) high and moderate confidence RAI-R CAU patients versus NON-R CAU patients (n RAI-R,CAU = 244 vs. n NON-R,CAU = 901), and (iv) high and moderate confidence RAI-R CAU and AA patients versus NON-R CAU and AA patients (n RAI-R,CAU + AA = 253 vs. n NON-R,CAU + AA = 947). Germline SNPs were selected from the candidate gene association analyses by applying the following filtering criteria: (i) apply p < 0.01 significance threshold and filter out SNPs with a minor allele frequency >10%, (ii) remove germline variants not unique to RAI-R patients except for variants that have a p < 0.01 statistical significance and an odds ratio >2, and (iii) remove germline variants that were not replicated in the four independent association analyses.
For the GWAS, both AA and CAU patients were included, regardless of their ancestry proportions, and the association was performed using both high and moderate confidence RAI-R patients (n RAI-R = 253 vs. n NON-R = 1010). Variants associated with RAI-R CAU patients have yet to be further analyzed. To identify variants strongly associated with African ancestry, all variants with a minor allele frequency of >1% in any population except the 1000 Genome African super-population were filtered out. Candidate loci were selected using the following criteria: (i) identify reference SNPs, those with the most significant association, by filtering out all germline variants from the GWAS using a significance threshold of p < 0.001, (ii) evaluate LD structure in the reference SNP by generating LD association plots for all SNPs (p < 0.001) within a genomic region ±100 kb of each reference SNP, (iii) remove loci that do not have ≥20 SNPs within the queried region, and (iv) remove loci that do not have ≥10 SNPs with LD D′ >0.6 of the reference SNP.
PCR-Seq analysis to verify imputed candidate RAI-R risk alleles and tumor driver mutations in AA thyroid cancer patients
PCR-Seq was performed to verify candidate RAI-R risk alleles in patients' blood DNA (OSU n = 55, MDACC n = 14, and NCI n = 7), and to detect BRAFV600E (17), NRASQ61R (18), and HRASQ61R (19) somatic mutations from available formalin-fixed paraffin-embedded (FFPE) tumors of AA thyroid cancer patients (OSU 28/55).
Statistical analysis
Logistic regression was applied to test the association of germline genetic variants with RAI-R disease. Multivariable analysis adjusting for age, sex, and race/ethnicity were performed. Haplotype estimation using observed genotypes was conducted with PHASE v2.1.1 (20). The chi-square test or Fisher's exact test was used to compare categorical variables, and the t-test or nonparametric Wilcoxon's test was used to compare continuous variable between groups. In order to identify any familial relationships among patient samples, identity by descent estimates or proportions (IBD Pi-HAT) for all pairs of individuals were obtained with PLINK (16) software. Pathogenicity of candidate variants was predicted with SIFT, PolyPhen, and Mutation Taster using ANNOVAR software.
Results
TG, BRCA1, and NSMCE2 haplotypes are uniquely associated with RAI-R disease in AA patients with >80% African ancestry
The candidate gene association analyses in select DNA-damage repair and iodine metabolism genes yielded 69 SNPs from 17 genes associated with RAI-R disease from CAU and/or AA patients (see Supplementary Table S1, which also includes 23 SNPs of the NSMCE2 risk haplotype identified by GWAS). GWAS generated 11 candidate loci associated with RAI-R disease in AA patients. Five candidate loci were comprised of SNPs located within the exons/introns of genes involved in DNA-damage repair, cell-cycle regulation, and immune response. These included: 6q22.33 (THEMIS), 8q24.13 (NSMCE2), 12q21.31 (ALX1), 17q21.31 (BRCA1, NBR1, NBR2), and 22q13.2 (EFCAB6). The remaining six candidate loci were comprised of SNPs located in intergenic regions and included: 6q13, 9q22.32, 9q31.1, 10q23.31, 12p13.31, and 12q21.32. Among the candidate variants, it was noted that thyroglobulin (TG), breast cancer type I susceptibility protein (BRCA1), and non-structural maintenance of chromosomes (SMC) element 2 (NSMCE2, also called MMS21) risk haplotypes were uniquely associated with 7/7 RAI-R AA patients of >80% African ancestry. A summary of the demographics of the OSU cohort of AA patients with >80% African ancestry is provided in Supplementary Table S2. None of the 30 NON-R AA patients of >80% African ancestry had residual or recurrent disease following 131I treatment.
The TG risk haplotype (p = 0.00087) includes eight SNPs with intermediate LD (R 2 > 0.60, D′ > 0.66 among all SNPs). All SNPs are located in TG introns 41–45, a region that has been reported to be predisposing to PTC (21). The BRCA1 risk haplotype (p = 0.00562) includes 14 SNPs in high LD (R 2 > 0.89, D′ > 0.90 among all SNPs). The SNP rs56082113 is a missense variant (NM_007294.3, BRCA1_K820E) located in exon 11. This variant has been reported in AA patients with high-risk breast cancer (22) or triple-negative breast cancer (23), although this variant has been reported in ClinVar to be likely benign in breast and ovarian cancer risk. The NSMCE2 risk haplotype (p = 0.00087) includes 23 SNPs in intermediate LD (R 2 > 0.38, D′ > 0.92 among all SNPs). The SNP rs150168649 is a missense variant (NSMCE2_F126C) predicted to be pathogenic by SIFT, PolyPhen, and MutationTaster. The estimated risk alleles for the TG, BRCA1, and NSMCE2 haplotypes are shown in Figure 2.
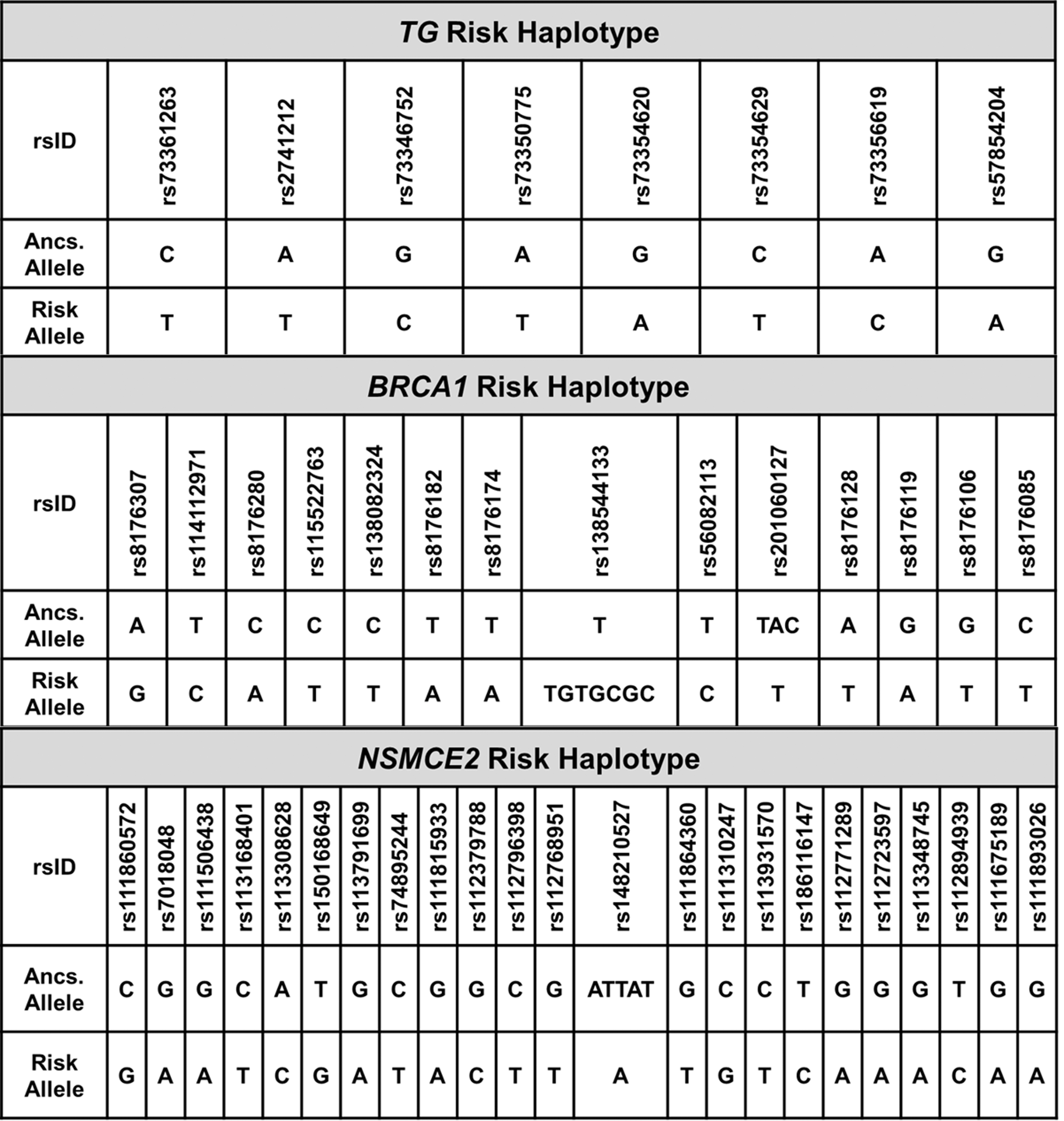
Estimated risk alleles for TG, BRCA1, and NSMCE2 haplotypes. Haplotype estimation of the risk alleles for the TG, BRCA1, and NSMCE2 haplotype using the observed genotypes was conducted with PHASE v2.1.1. Ancs. Allele: ancestral allele.
The frequencies of the risk alleles were calculated for the SNPs comprising the TG, BRCA1, and NSMCE2 estimated RAI-R haplotypes for OSU AA patients of >80% African ancestry (n RAI-R = 7, n NON-R = 30), and 1000 Genome European and African super-populations (Fig. 3). Risk allele frequencies are enriched in RAI-R AA patients of >80% African ancestry.
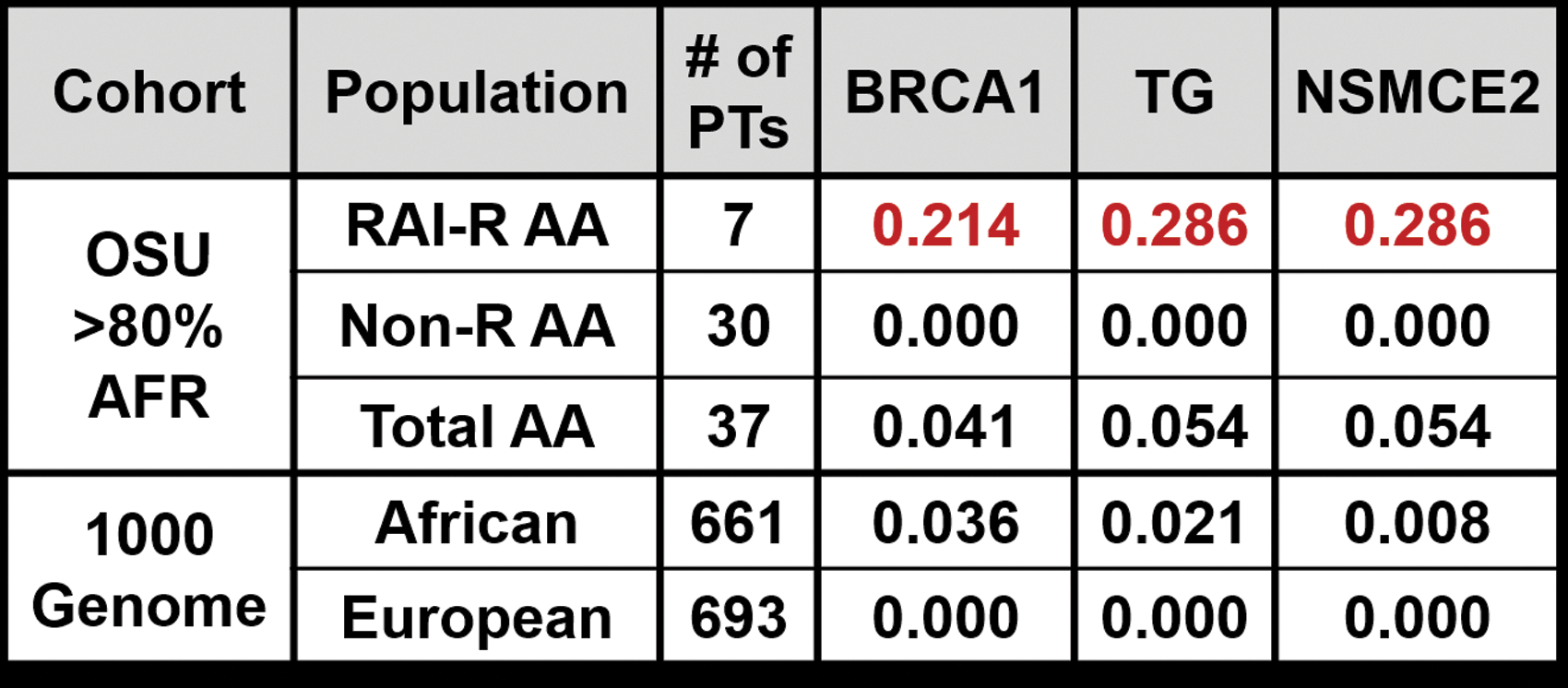
TG, BRCA1, and NSMCE2 risk allele frequencies. Risk allele frequencies were calculated for the TG, BRCA1, and NSMCE2 candidate RAI-R haplotypes using the observed genotype counts for AA patients with >80% African ancestry in the Ohio State University (OSU) cohort, and compared to the reported allele frequencies for candidate variants in the African and European super-populations in the 1000 Genome Phase 3 data. PTs, patients; NON-R, non-radioiodine refractory.
As shown in Figure 4, it was noted that (i) the TG haplotype was uniquely identified in four RAI-R AA patients, all of whom had elevated serum Tg levels after RAI therapy; (ii) the NSMCE2 haplotype was uniquely identified in four RAI-R AA patients, all of whom were diagnosed at an early age (13, 17, 17, and 26 years old) with distant metastatic disease at initial diagnosis; and (iii) the BRCA1 haplotype co-occurred with the NSMCE2 haplotype in three out of four patients. The SNPs comprising the TG, NSMCE2, and BRCA1 haplotypes were not detected in any other patients in the cohort. As SNPs comprising the risk haplotypes were derived from imputation, Sanger sequencing was performed, which verified the presence and accuracy of the candidate RAI-R risk alleles. In addition, Sanger sequencing demonstrated that none of the TG, BRCA1, or NSMCE2 haplotypes were detected among self-reported AA patients with an unknown proportion of African ancestry from MDACC (n RAI-R = 5, n NON-R = 9) or the NCI (n RAI-R = 1, n NON-R = 6).
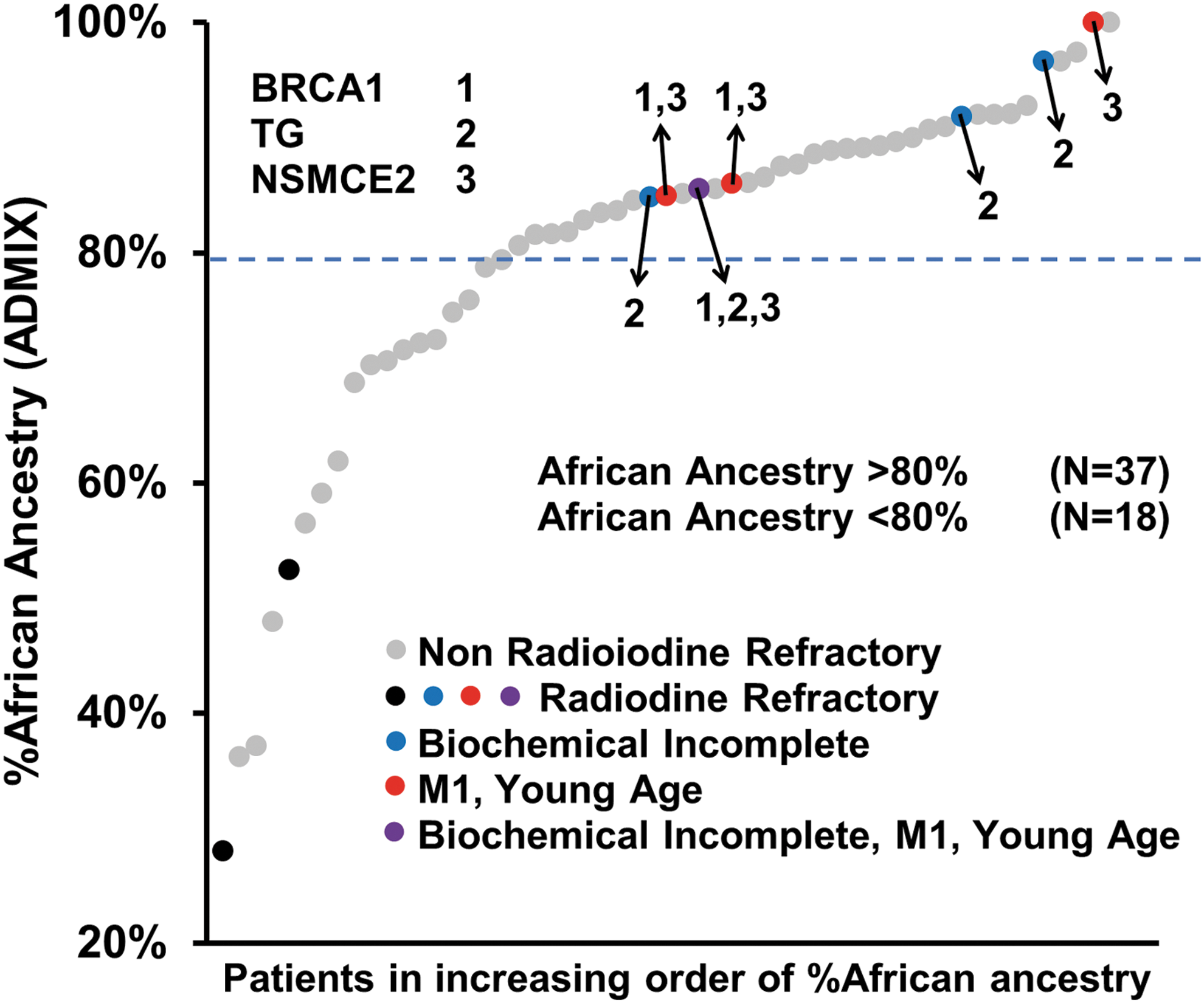
Association of candidate RAI-R haplotypes with AdMixture African ancestry in the OSU AA cohort. Each point along the x-axis represents a patient sample that is organized, from left to right, by increasing % African ancestry (y-axis). Genetic ancestry was calculated for all patients using the ADMIXTURE software. Per American Thyroid Association guidelines, a biochemical incomplete response was defined as basal serum thyroglobulin (Tg) >2 ng/mL or stimulated serum Tg >10 ng/mL. Young age includes patients 13–26 years of age. The TG, BRCA1, and NSMCE2 candidate RAI-R haplotypes, denoted as 2, 1, and 3, respectively, occurred in AA RAI-R patients, all of whom were of >80% African ancestry (7/9; 78%). Note that the BRCA1 haplotype co-occurred in three of four patients with the NSMCE2 haplotype, and one patient had a co-occurrence of the TG, BRCA1, and NSMCE2 haplotypes. M1, distant metastatic disease.
Incidence of BRAFV600E appears lower in AA patients of >80% African ancestry
In predominately CAU cohorts, BRAFV600E is the most common somatic variant in PTC (60%) (24). The incidence of BRAFV600E remains high in PDTC (30%) (25) and ATC (40%) (25,26). Among 28 AA patients who had FFPE tumors available for detection of somatic variants, 23/28 were PTCs and 5/28 were FTC. In this cohort, the overall incidence of BRAFV600E was 39.1% (9/23) for AA PTC patients and 0% (0/5) for AA FTC patients. As shown in Figure 5, the incidence of BRAFV600E in AA PTC patients of >80% African ancestry was 21% (3/14) compared to 67% (6/9) in AA PTC patients with <80% African ancestry (p = 0.077). NRASQ61R was observed in 14% (2/14) of AA PTC patients of >80% African ancestry, and in 50% (1/2) of AA FTC patients of <80% African ancestry. HRASQ61R was unobserved in these 28 AA patients. Of interest, the primary tumors available from three RAI-R AA patients of >80% African ancestry were negative for BRAFV600E , NRASQ61R , and HRASQ61R mutations, suggesting alternative tumor driver mutations for these three RAI-R AA patients.

Incidence of BRAFV600E , NRASQ61R , and HRASQ61R tumor driver mutations in primary tumors available from OSU AA patients. Each point along the x-axis represents a patient sample that is organized, from left to right, by increasing % African ancestry (y-axis). Sanger sequencing was performed on available AA formalin-fixed paraffin-embedded primary tumor samples (28/55) to identify the incidence of the three most frequently reported somatic mutations in thyroid cancer: BRAFV600E , NRASQ61R , and HRASQ61R . Among 28 tumors, 23 were papillary thyroid carcinomas (PTC, black) while the remaining five tumors were follicular thyroid carcinomas (FTC, light gray). HRASQ61R , not shown in the inset table, was not detected in any of the 28 primary tumor samples. For the inset table, PTC (n = 23) includes classic PTC, micro PTC (mPTC), and follicular variant PTC (FvPTC).
Discussion
This study identified three candidate risk haplotypes involving the TG, BRCA1, and NSMCE2 genes, uniquely found in RAI-R patients of >80% African ancestry. The TG haplotype was associated with persistently elevated serum Tg after RAI therapy, and the BRCA1 and NSMCE2 haplotypes were associated with diagnosis at a young age and distant metastasis. Furthermore, the primary tumors of RAI-R patients of >80% African ancestry (n = 3) were negative for the most common somatic variants detected in PTCs. While germline variants predisposing thyroid cancer have been reported, this is the first investigation of germline variants associated with RAI-R disease. Finally, the data suggest a differential genetic landscape underlying thyroid cancer between AA and CAU patients.
Germline variants and thyroid cancer
Thyroid cancer has been reported to have a strong genetic component, as evidenced by its familial heritability (27,28). Both GWAS and candidate gene association studies have reported strong associations between SNPs located within the 2q35 (DIRC3), 8p12 (NRG1), 9q22.33 (FOXE1), and 14q13.3 (NKX2-1) loci and thyroid cancer predisposition in cohorts composed of CAU and Asian populations (13,29 –31). Jendrzejewski et al. (32) reported that germline variants mapped to the 14q13 locus are associated with specific histological subtypes of PTC. However, germline variants associated with tumor progression and/or 131I therapeutic response of thyroid cancer have not been investigated. To date, studies of thyroid cancer have focused on the tumor mutation landscape associated with different stages of disease, which is of prognostic value and provides actionable targets for therapeutic intervention. However, germline variants influencing pharmacokinetics and pharmacodynamics are well recognized (33), and several germline pharmacogenetics variants are Food and Drug Administration approved to guide anticancer treatments (34,35). Likewise, genetic variants have been identified in the DNA-damage repair genes ATM (36), NBS1, MRE11, and LIG4 that lead to increased radiosensitivity, which results in severe toxicity in normal tissues and the development of second malignancy after external radiation (36,37). Genetic variants associated with radioresistance, likely a complex trait, have not been reported. Patients' refractoriness to RAI therapy may result from insufficient RAI delivery and/or RAI resistance. The interaction between tumor mutation landscape and genetic variants influencing the inflammatory microenvironment and systemic immune surveillance of neoantigens exposed by tumor killing likely contribute to RAI resistance. For RAI delivery, BRAF/MEK inhibitors have been shown to restore/enhance RAI delivery in metastatic lesions of some patients carrying tumor driver mutations that lead to overactivation of the MAPK pathway (7). Genetic variants influencing the pharmacokinetics of BRAF/MEK inhibition may contribute to differential responsiveness to this class of inhibitors among individual patients. Taken together, the integration of tumor mutation landscape with germline variants associated with response to RAI therapy will predict or stratify RAI-R disease into distinct phenotypes to allow the selection of optimal treatments for each patient.
TG, BRCA1, and NSMCE2 risk haplotypes
The primary function of Tg is to provide the scaffold for iodination of selected tyrosine residues within the protein to produce thyroid hormone. Accordingly, Tg plays an important role in RAI retention and thus absorbed 131I activity in target lesions. The candidate TG haplotype includes eight SNPs located in TG introns 41–45, all of which are variants of unknown significance. Of interest, six out of eight of the candidate SNPs co-occur in TG intron 41, overlapping the protein-coding gene Src-like adaptor (SLA) and several long noncoding RNAs (lncRNAs) encoded by the opposite strand, in a previously identified 8q24 PTC susceptibility locus (21). The possible functions of SNPs in TG, SLA, and/or lncRNAs are not immediately apparent, and future studies discerning whether the candidate TG haplotype is merely a reporter or functionally contributes to the RAI-R phenotype are necessary.
The BRCA1 gene has been shown to have cell cycle–dependent expression (38), regulatory control of the G1-S and G2-M transitions (39,40), and involvement in DNA damage-repair processes, including homologous recombination, non-homologous end joining, and transcription-coupled repair (41). The candidate BRCA1 haplotype includes the missense variant rs56082113 BRCA1K820E , which is located in exon 11, within a DNase I hypersensitive site and region interacting with RNA Polymerase II (POLR2A). The potential role of the BRCA1 haplotype in RAI-resistance remains to be investigated.
NSMCE2 encodes the E3 SUMO-protein ligase component of the large SMC5–SMC6 complex, which is involved in the repair of DNA double-strand breaks via resolution of joint sister chromatid intermediates generated during homologous recombination. The Siz/PIAS Ring (SP-RING) domain, located in amino acids 154–236, is a putative zinc-finger binding domain that contains five conserved cysteine/histidine residues (C169, C185, H187, C210, and C215). Mutation or deletion of the SP-RING domain compromises DNA-damage response (42). The NSMCE2 haplotype includes the SNP rs150168649, a missense mutation (F126C) predicted to be pathogenic. NSMCE2F126C is in the proximity of the SP-RING domain and may compromise NSMCE2 function, leading to genomic instability and contributing to the RAI-R phenotype.
Both BRCA1 and NSMCE2 play important roles in DNA-damage repair. The co-occurrence of the BRCA1 and NSMCE2 risk haplotypes in three out of four RAI-R AA patients who were diagnosed at a young age with distant metastasis suggests that hypomorphic SNPs in multiple DNA damage repair genes may cumulatively contribute to RAI-R disease.
Tumor mutation landscapes for AA patients with thyroid cancer
For most cancer types, including thyroid cancer, AA have a higher cancer death rate than CAU (43 –45), most likely due to disparities in health care access and differences among their genetic landscapes. For example, in colorectal cancer, AA patients harbor a lower frequency of BRAF mutations than CAU patients (46 –48); and in melanoma, AA patients exhibit a decreased incidence of BRAF mutations (49). In The Cancer Genome Atlas PTC cohort, the incidence of BRAFV600E was 58% (15/26) in AA patients compared to 65% (209/321) in CAU patients (p = 0.236). In the Genie data set (50), the incidence of BRAFV600E was 30% (6/20) in AA PTC patients and 60% (208/346) in CAU PTC patients (p = 0.010). Of note, race/ethnicity of the AA patients in the aforementioned studies was self-reported, and the proportion of African ancestry was not determined using ancestry informative markers. Thus, there may be a greater disparity in tumor mutation signatures in AA patients of high African ancestry. In the present cohort of AA PTC patients, with African ancestry ranging from 28% to 100%, the incidence of BRAFV600E was 35% (8/23). For AA PTC patients with >80% African ancestry, the incidence of BRAFV600E was 21% (3/14). Altogether, the incidence of BRAFV600E appears to be lower in AA patients with colorectal cancer, melanoma, and thyroid cancer. These findings suggest that AA patients of high African ancestry may have distinct, potentially novel, tumor driver mutations that may partially contribute to their higher mortality. Further investigation of the tumor mutation landscapes at different stages of thyroid cancer for AA patients is warranted.
Validation of risk haplotypes and future directions
While the present results appear compelling, they must be interpreted with caution before a validation study is conducted in a larger cohort. After the association analyses were corrected for multiple testing via the Bonferroni procedure (51,52), the small sample number of the cohort prevents candidate SNPs from reaching appropriately adjusted statistical significance thresholds. The probability of population stratification bias is increased, as AA patients represent 4.6% (55/1200) of our entire cohort and 3.6% (9/253) of the RAI-R cases. Statistical modeling of genetic association studies indicate that (i) the larger the differences in either allele frequency or disease prevalence between the subgroups existing in a study population, the greater the type I error, and (ii) the type I error rate is greater for uncommon variants (i.e., variants with a minor allele frequency of <5%) (53). In the present cohort, the difference in RAI-R disease prevalence between CAU (21.3%) and AA (16.4%) patients is 4.9%, while the allele frequencies for the TG, BRCA1, and NSMCE2 candidate RAI-R haplotypes are 0% for RAI-R CAU patients and 24.1–28.6% for RAI-R AA patients. Additionally, the individual SNPs comprising each of the TG, BRCA1, and NSMCE2 candidate RAI-R haplotypes are rare variants, as their frequencies are between 0.8% and 3.6% in the 1000 Genome African super-population.
Attempts to validate candidate RAI-R risk haplotypes in additional AA patients are challenging, and several hurdles need to be overcome. First, the institutes need to have IRB-approved protocols for bio-repositories with sufficient clinical follow-up to determine RAI-R status for their patients. Second, it is difficult to target AA patients of >80% African ancestry, as the percentage of African ancestry is typically unknown and clearly varies among self-reported AA patients. The IBD analysis (IBD Pi-HAT, <0.05) indicates that the AA patients with risk haplotypes in the cohort are unrelated. In addition to pursuing validation in a larger cohort of AA patients, functional annotation of the TG, BRCA1 and NSMCE2 variants will be pursued in search of molecular explanations for their association with RAI-R disease.
Conclusion
Racial/ethnic differences in cancer incidence rates, aggressiveness, histopathological features, patient outcome, and germline/somatic mutation frequencies have often been reported across multiple cancer types. However, the causes of reported racial/ethnic disparities remain poorly understood and understudied. To this end, the germline genetic landscape of AA thyroid cancer patients were investigated, and candidate TG, BRCA1, and NSMCE2 RAI-R risk haplotypes occurring uniquely in AA patients of >80% African ancestry were identified. Continued study of candidate RAI-R germline SNPs may help to identify AA patients at risk of developing RAI-R disease for strategic intervention with combination therapy using small molecule inhibitors or immunotherapy. Further investigation of the functional annotation of these germline SNPs could provide valuable insights into the mechanisms underlying RAI-R disease.
Footnotes
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) Grant P01CA124570 (Project 1 leader: A. de la Chapelle, Project 3 leader: S.M. Jhiang; PI: M.D. Ringel), NIH Grant P50CA168505 (Project 1 co-leader: A. de la Chapelle, Project 2 co-leader; S.M. Jhiang; PI: M.D. Ringel).
Author Disclosure Statement
The authors have nothing to disclose. No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2