
Abstract
Background:
Prior studies have reported mutations in mismatch repair (MMR) genes in a small subset of anaplastic thyroid carcinomas (ATC). The aim of this study was to identify MMR-protein-deficient (MMR-D) ATC and investigate their histopathologic features and clinical outcome.
Methods:
A cohort of 28 ATC diagnosed between 2003 and 2017 with tissue blocks available were evaluated. Immunohistochemistry for MMR proteins was performed to identify MMR-D tumors. Clinicopathologic features, molecular findings (determined by a targeted next-generation sequencing assay), and clinical outcome data for MMR-D tumors were recorded and compared to that of MMR-protein-intact (MMR-I) tumors.
Results:
There were four (14%) MMR-D ATC, all of which showed complete loss of MSH2 and MSH6 with intact expression of MLH1 and PMS2. Three of these tumors had MSH2 mutations and a hypermutated phenotype by next-generation sequencing. All four patients (two male; M age at diagnosis = 64 years) presented with stage IVB disease (i.e., gross extrathyroidal extension or a lymph node metastasis at presentation). There were no differences in tumor size or rates of gross extrathyroidal extension, lymph node metastases, or positive resection margins between MMR-D and MMR-I ATC. Patients with MMR-D tumors were less likely to have distant metastatic disease at presentation (p = 0.035), although half did eventually develop distant metastases. MMR-D tumors were not histologically distinct. All four patients with MMR-D tumors lived for more than one year. One patient died of disease at 15 months, while the remaining three were alive at last follow-up, with survival of 19, 38, and 48 months. Patients with MMR-D ATC had significantly better survival compared to those with MMR-I tumors (p = 0.033), which was maintained when considering only patients with stage IVB disease at presentation (p = 0.030).
Conclusion:
MMR-D tumors comprised 14% of this ATC cohort. Although the findings must be interpreted with caution given the small number of MMR-D ATC in the cohort, the results suggest that MMR status may be prognostically significant in ATC.
Introduction
Anaplastic thyroid carcinoma (ATC) is a rare, aggressive tumor that accounts for <5% of all thyroid carcinomas but is responsible for a significant subset of thyroid cancer-related deaths (1), with a median survival of approximately 4–12 months (2 –5). Given their poor prognosis, all ATC are categorized by the American Joint Committee on Cancer (AJCC) as stage group IV (IVA, intrathyroidal; IVB, gross extrathyroidal extension or lymph node metastasis; or IVC, distant metastasis) (6). Almost all ATC patients die of their disease. However, improved survival has been associated with lower stage at diagnosis, surgical resection, and multimodal therapy (2 –5,7). ATC are genetically heterogeneous. Although some ATC may arise de novo, a significant subset harbor BRAF and RAS mutations, consistent with the idea that many arise from more differentiated thyroid carcinomas (5,8 –10). ATC have a higher mutational burden compared to differentiated thyroid carcinomas and frequently harbor TP53, TERT, and PIK3CA mutations, among other alterations (5,8 –10). Mutations in genes involved in the mismatch repair (MMR) pathway have also been implicated in the tumorigenesis of a small subset of ATC (9,10).
MMR deficiency is associated with a hypermutated phenotype characterized by an elevated number of single nucleotide variants (SNVs), as well as increased numbers of small insertions/deletions (indels) within DNA repeat regions, a characteristic phenomenon termed “microsatellite instability” (MSI). MMR deficiency may be caused by several mechanisms, including inactivating mutations within MMR genes (i.e., MLH1, PMS2, MSH2, and MSH6), MLH1 promoter hypermethylation, and EPCAM deletion with subsequent MSH2 promoter hypermethylation (11 –13). In the germline setting, MMR mutations result in an increased risk for developing malignant neoplasms (Lynch syndrome). Although MMR deficiency occurs in a significant subset of some cancer types, including approximately 15% of colorectal carcinomas and approaching 30% of endometrial carcinomas, it is not known to occur in differentiated thyroid carcinomas (14). Mutations in MMR genes have been reported in approximately 10% of ATC (9,10). However, the characteristics of these ATC have not been established. The aim of this study was to utilize immunohistochemistry (IHC) for MMR proteins to identify MMR-protein-deficient (MMR-D) ATC and evaluate their histopathologic features and clinical outcome.
Methods
Study population and data acquisition
Approval from the Brigham and Women's Hospital Institutional Review Board was obtained. Cases of ATC were identified that were diagnosed from January 2003 to August 2017 and had paraffin-embedded tissue blocks available. For all cases that met inclusion criteria, slides were reviewed to confirm the diagnosis, and histopathologic features (tumor morphology, presence of a more differentiated precursor lesion, and proportion of the anaplastic component) were recorded. A total of 37 ATC were identified. Following histopathologic review, nine tumors that demonstrated only focal progression to ATC (≤10% of the tumor cellularity) were excluded because previous studies have shown that patients with tumors with only a minor anaplastic component (or “incidental” ATC in an otherwise differentiated thyroid carcinoma) have a prolonged survival compared to patients with tumors with a major anaplastic component (15 –20). For tumors that were resected, additional characteristics (tumor size, margin status, presence of lymphovascular invasion or extrathyroidal extension, and lymph node status) were also documented. For each patient, demographic information (age and sex), treatment regimen, and clinical outcome information were obtained from the electronic medical record.
IHC evaluation of MMR-D
IHC was performed on formalin-fixed paraffin-embedded tissue sections 4 μm thick using mouse anti-MLH1 monoclonal antibody (1:100 dilution; clone ES05; Novocastra, Buffalo Grove, IL), mouse anti-PMS2 monoclonal antibody (1:50 dilution; clone MRQ-28; Cell Marque, Rocklin, CA), mouse anti-MSH2 monoclonal antibody (1:150 dilution, clone FE11; Calbiochem, San Diego, CA), and mouse anti-MSH6 monoclonal antibody (1:50 dilution, clone PU29; Novocastra) using the Envision Plus Detection System (Dako, Carpinteria, CA). Cases were characterized as MMR-D if tumor cells showed loss of staining for any of the four proteins, with intact staining in adjacent tissue (i.e., an internal positive control was required).
Molecular characterization by targeted next-generation sequencing
A subset of patients had molecular characterization of their tumors as part of Profile:OncoPanel using a targeted next-generation sequencing (NGS) assay developed at Brigham and Women's Hospital (21,22). This included all four MMR-D cases. OncoPanel molecular testing was performed using formalin-fixed paraffin-embedded tissue or freshly frozen tissue. Tumor DNA was isolated per standard methods (Qiagen, Valencia, CA) from macro-dissected regions of the tumor. Libraries were prepared from 50 ng DNA using a customized solution-phase hybrid capture approach (Agilent Technologies, Santa Clara, CA). Depending on the version of OncoPanel used at the time, bait sets covered either 298 genes or an extended panel of 447 genes. NGS was performed using an Illumina HiSeq 2500 (Illumina, San Diego, CA). Sequencing was analyzed by a variety of internally developed and publicly available tools, as previously described (22).
Evaluation of microsatellite instability
Microsatellite instability was evaluated in one case (the one MMR-D case that lacked alterations in all four MMR genes and lacked an overt hypermutated phenotype by NGS). This was performed by polymerase chain reaction (PCR) amplification across five microsatellite loci (four mononucleotide repeats [BAT25, BAT26, BAT40, and BAT34c] and one dinucleotide repeat [D18S55]) with fluorescently labeled primers in matched tumor and normal DNA samples. PCR products were analyzed using capillary gel electrophoresis (3130x1 Genetic Analyzer; Applied Biosystems, Foster City, CA).
Statistical analysis
A t-test or Fisher's exact test was used to compare differences in continuous and categorical variables, respectively. Overall survival was depicted using the Kaplan–Meier method, and comparisons between subgroups were performed using the log-rank test. For all statistical methods, p-values <0.05 were considered significant.
Results
Clinicopathologic characteristics of the entire cohort
A total of 28 cases of ATC were identified that met the inclusion criteria. This included tumors from 13 (46%) women and 15 (54%) men, with a mean age of 67 years at the time of diagnosis (range 41–85 years). Two (7%) patients presented with disease confined to the thyroid (stage IVA), while 11 (39%) had gross extrathyroidal extension or lymph node metastases (stage IVB) and 15 (54%) had distant metastases at diagnosis (stage IVC). On pathologic examination, the mean size of resected tumors was 6.3 cm (range 2.0–10.0 cm), 10 (63%) had gross extrathyroidal extension, and the resection margins were positive in nine (56%) cases. Lymph nodes were sampled in 13 (81%) cases, with seven (54%) demonstrating metastases. Twenty-three (82%) patients had thyroid tumors that were initially diagnosed as ATC, while five (18%) patients had a history of a more differentiated thyroid carcinoma that recurred as ATC. Of patients who were diagnosed with ATC at presentation, 10 (43%) underwent initial total or near-total thyroidectomy, six (26%) had a lobectomy/hemithyroidectomy, and seven (30%) had a biopsy only. Of the 26 patients with detailed treatment history available in the medical record, three (12%) had additional radioactive iodine, 20 (77%) had external radiation therapy, 13 (50%) had chemotherapy, and seven (27%) had targeted therapy. Overall, 22 (79%) patients died of disease. Of the 28 patients, 10 (36%) survived for at least one year. Excluding one patient without sufficient follow-up, seven (26%) survived for at least two years. Of those who died of disease, the median survival was five months (M = 8 months; range 1–44 months).
MMR-D ATC
Of the 28 ATC in the cohort, four (14%) demonstrated MMR-D by IHC, each showing complete loss of MSH2 and MSH6 nuclear expression within tumor cells (Fig. 1). Clinicopathologic features of the four MMR-D ATC compared to the 24 MMR-protein-intact (MMR-I) ATC are summarized in Table 1. The four patients with MMR-D tumors included two (50%) women and two (50%) men, with a mean age of 64 years (range 51–84 years). All four patients lacked a personal or family history suggestive of Lynch syndrome. All patients presented with stage IVB disease. Two (50%) patients underwent initial total thyroidectomy and two (50%) a hemi-thyroidectomy. The mean size of MMR-D tumors was 5.8 cm (range 3.0–8.0 cm), three (75%) had significant gross extrathyroidal extension, and the resection margins were positive in two (50%) cases. Lymph nodes were sampled in three (75%) cases, with metastatic disease in one (33%) case. There were no differences in tumor size or rates of gross extrathyroidal extension, lymph node metastases, or positive resection margins between MMR-D and MMR-I ATC. Patients with MMR-D tumors were less likely to have distant metastases at presentation (p = 0.035), although half did eventually develop distant disease.

Immunohistochemistry (IHC) for mismatch repair (MMR) proteins. This anaplastic thyroid carcinoma (ATC) harbored an MSH2 mutation and had a hypermutated phenotype. The tumor showed intact expression of MLH1 (
Clinicopathologic Characteristics of MMR-D vs. MMR-I ATC
Of patients with first-time thyroid cancer diagnoses.
Of patients who received adjuvant therapy with detailed treatment history.
Excluding one patient without long enough follow-up.
Of patients who died of disease.
Of patients who had surgical resection.
Of patients with lymph node sampling.
ATC, anaplastic thyroid carcinoma; MMR-D, mismatch repair-protein-deficient; MMR-I, mismatch repair-protein-intact.
MMR-D tumors were not morphologically distinct. The morphology of the MMR-D tumors was heterogeneous, with combinations of epithelioid, spindled, and/or giant cell components (Fig. 2). Four (100%) MMR-D tumors demonstrated areas of epithelioid morphology, two (50%) had areas with a spindled morphology, none had areas with a squamoid morphology, and one (25%) had areas with a giant cell morphology compared to 13 (54%), 17 (71%), five (21%), and three (13%) for MMR-I tumors, respectively (p > 0.05 for each morphologic comparison). All MMR-D ATC were composed predominantly of ATC. Three cases (75%) had a marked peritumoral inflammatory infiltrate compared to 46% of MMR-I ATC (p = 0.35). None of the four cases had a well-differentiated thyroid carcinoma component or a prior history of a differentiated thyroid carcinoma (in contrast to 38% of MMR-I cases; p = 0.27). However, three (75%) demonstrated a more monomorphic component with a clear cell or oncocytic cytomorphology suggestive of a component of poorly differentiated thyroid carcinoma. Molecular characterization of the four MMR-D tumors demonstrated that all cases lacked a BRAF or RAS mutation. Three (75%) of the MMR-D tumors contained inactivating MSH2 mutations (p.T441mfs*13, p.P622Qfs*13, and p.H783Qfs*31). All three of these cases had a hypermutated phenotype, with tumor mutational burdens (TMB) of 19.01, 25.27, and 28.14 mutations/mb. The final case lacked alterations in all four MMR genes and additionally lacked an EPCAM deletion. Although it did not exhibit an overt hypermutated phenotype (defined by a TMB cutoff or 12) (23), it was found to have mutations in TP53 and NF1, as well as within a few homopolymer repeat regions, and had a TMB of 10.83 mutations/mb. MSI testing of this tumor revealed that it was microsatellite stable (MSS).
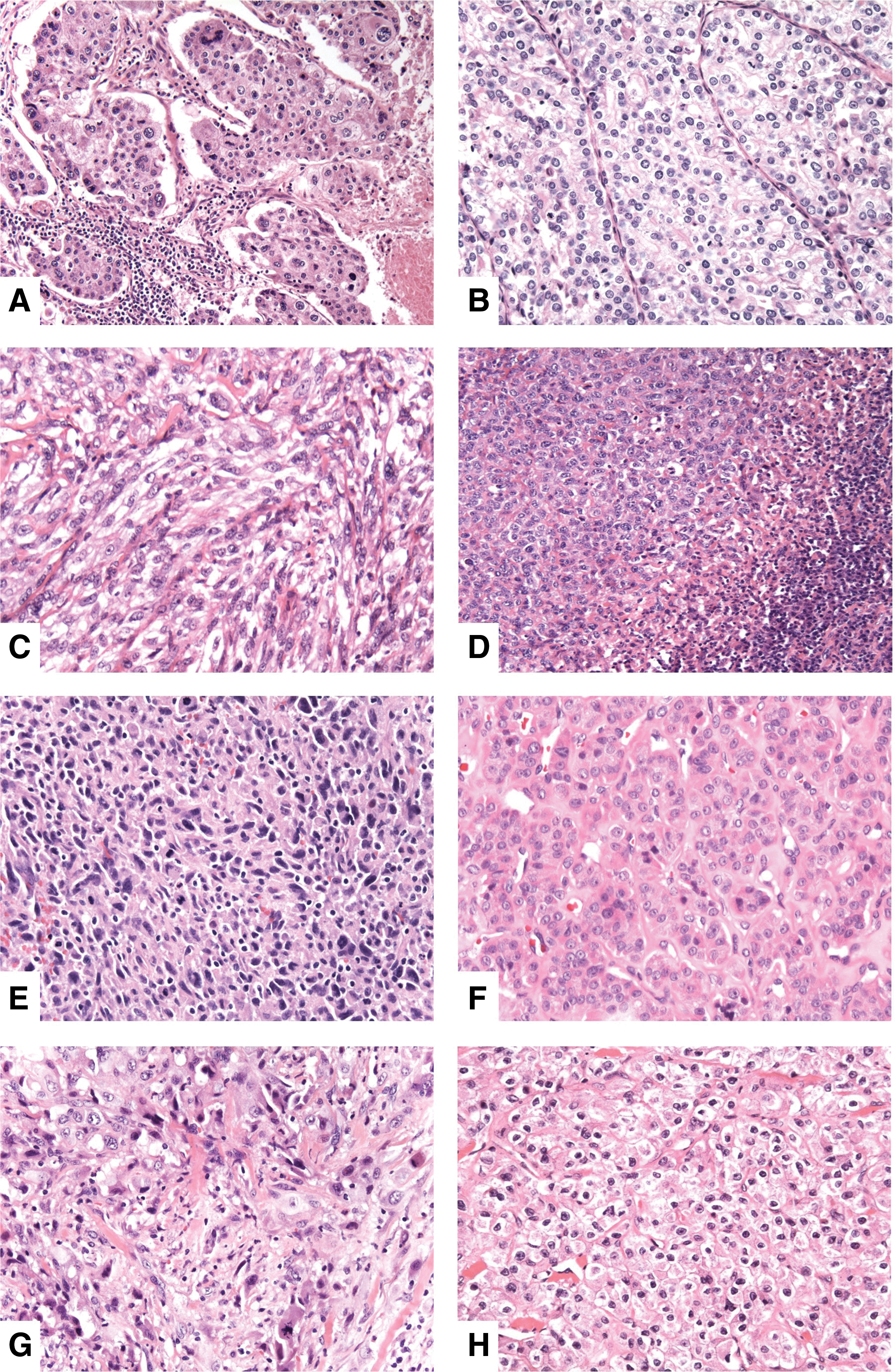
Morphology of mismatch repair-protein-deficient (MMR-D) ATC. (
All four patients with MMR-D ATC had adjuvant external radiation therapy and chemotherapy. Two (50%) patients developed distant metastases to the lung after initial presentation, while the other two had no evidence of recurrence at last follow-up. All patients survived for at least one year. One patient died of disease at 15 months, while the remaining three were alive at last follow-up with survival of 19, 38, and 48 months. Whereas MMR-D ATC comprised 14% of the overall cohort, they accounted for 40% of cases with survival of one year or more. Patients with MMR-D ATC had a significantly better survival compared to patients with MMR-I tumors (p = 0.033), which was maintained when considering only those with stage IVB disease at presentation (p = 0.030) (Fig. 3). When comparing only patients with loss of MMR protein expression and a hypermutated phenotype to those with tumors with intact expression of MMR proteins, there was a trend toward better survival when considering all patients (p = 0.087) and those who presented with stage IVB disease (p = 0.070).
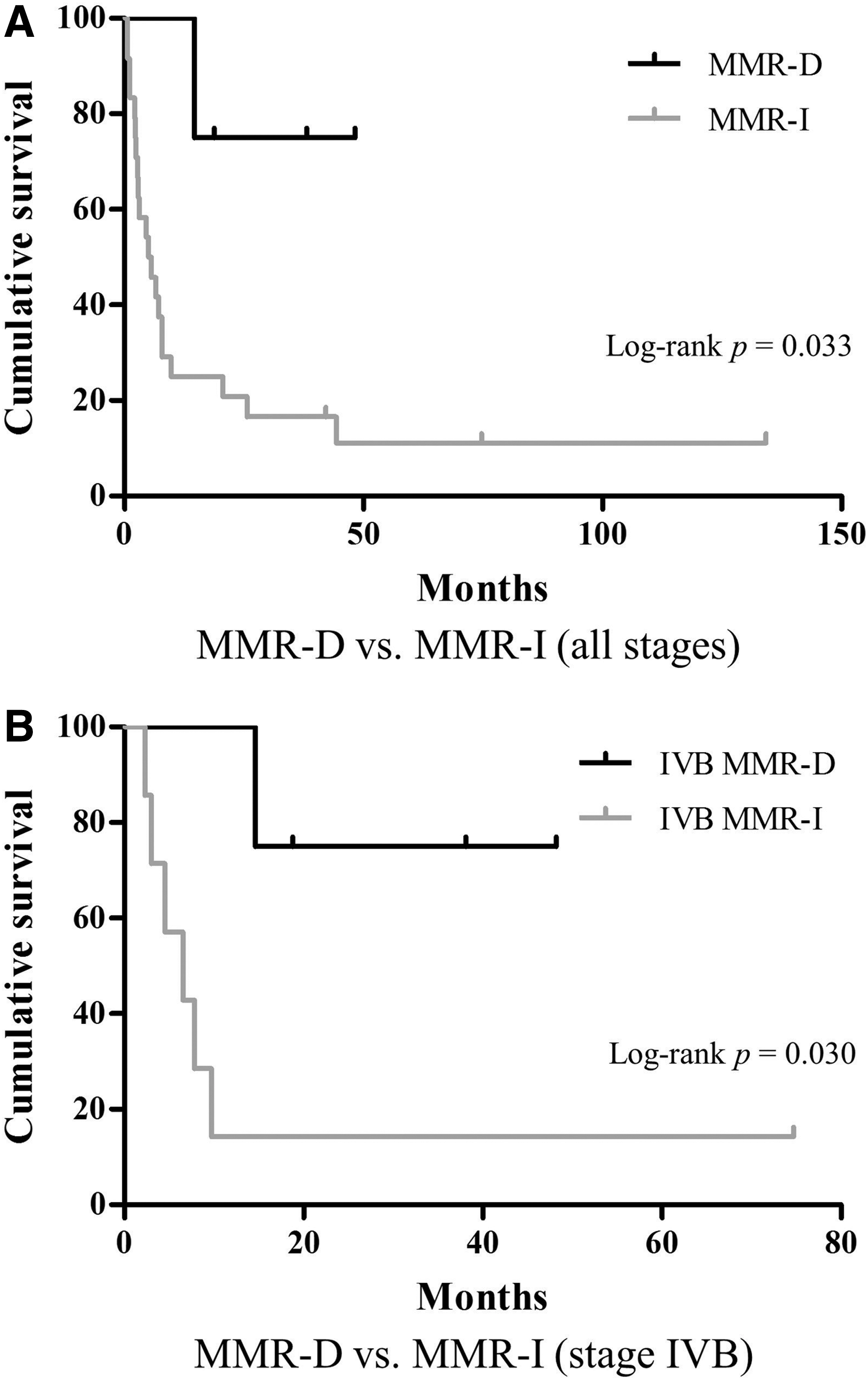
(
Discussion
The aim of this study was to identify MMR-D ATC and investigate their histopathologic features and clinical outcome. It was found that MMR-D ATC comprised 14% of the cohort, a rate consistent with the results of two prior studies that evaluated the genetic landscape of ATC (9,10). On histopathologic examination, MMR-D tumors were heterogeneous with variable epithelioid, spindled, and giant cell morphologies. None of the cases had a well-differentiated component, though three tumors had a more monomorphic component suggestive of poorly differentiated thyroid carcinoma. Three cases had a marked peritumoral inflammatory infiltrate. Overall, MMR-D tumors were not morphologically distinguishable from MMR-I ATC. Thus, the results indicate that similar to other organ systems, morphology alone cannot be used to identify MMR-D tumors (24 –26).
All four MMR-D ATC demonstrated loss of expression of MSH2 and MSH6. Three of the four cases were found to have an MSH2 mutation by targeted NGS. MSH2 mutations can be somatic or germline in the setting of Lynch syndrome. Although ATC has been reported in two patients with Lynch syndrome, it is thought to be an extremely rare association (27,28). Both of the reported Lynch syndrome–associated ATC demonstrated loss of MSH2 expression. Interestingly, both of these cases were found to be MSI-low (i.e., MSI testing showed instability at only 1/5 loci). Germline testing was not performed in this study. Although it was difficult to exclude the possibility that the MSH2 mutations were germline events based on the mutation allele frequencies, all four patients with MMR-D tumors lacked a personal or family history suggestive of Lynch syndrome. The explanation for the loss of MSH2 and MSH6 expression in the one case that lacked an MSH2 mutation is unclear. Deletions of the 3′ portion of EPCAM have been associated with MSH2 promoter hypermethylation with resulting loss of MSH2 expression (12). However, no EPCAM alteration was detected in the tumor. It is possible that a genetic alteration that is undetectable by the OncoPanel sequencing assay is leading to MSH2 inactivation in the tumor. Notably, inversions of exons 1–7 of MSH2 are a known cause of Lynch syndrome when present in the germline (29). A similar somatic alteration could exist in the tumor in this cohort. The concomitant loss of MSH6 expression supports the apparent functional inactivation of MSH2. Finally, while the tumor was MSS by PCR testing, OncoPanel analysis did identify several small insertions/deletions within homopolymer regions, potentially suggesting some degree of MMR deficiency. However, definitively addressing this question was beyond the scope of this study.
Patients with MMR-D ATC had a significantly improved survival compared to those with MMR-I tumors. Moreover, whereas MMR-D ATC comprised 14% of the overall cohort, they were enriched in tumors from patients surviving for a year or more, comprising 40% of such cases. All four patients with MMR-D ATC survived for more than one year and two survived for more than three years, despite the fact that all patients with MMR-D tumors presented with stage IVB disease. Significantly, the difference in survival was maintained when considering only patients with stage IVB disease at presentation. Although this finding must be interpreted with caution because of the small number of MMR-D ATC in the cohort, the finding is intriguing given that MMR deficiency has been linked to improved outcome in other tumors such as colorectal carcinoma, with improved survival thought to be a result of heightened antitumor immune response secondary to an increased neo-antigen burden produced as consequence of the hypermutated phenotype of MMR-D tumors (30,31). Outcome data were presented in one of the two studies that evaluated the molecular profile of ATC (9). In the study by Kunstman et al., both patients with ATC who harbored mutations in MMR genes had large tumors and developed distant metastases. One of the patients survived for almost 40 months, while the second patient died within three months, indicating that not all patients with MMR-D ATC have a prolonged survival.
In summary, MMR-D tumors comprised 14% of this ATC cohort, and patients with MMR-D ATC had an improved survival compared to those with MMR-I tumors. Increasingly, ATC patients are being treated with checkpoint inhibitor therapy both because there is currently no curative treatment for ATC and because there is emerging evidence that checkpoint inhibitor therapy may be efficacious for ATC patients (32,33). Given the known association between MMR status and response to checkpoint inhibitor therapy (34), determining MMR status may have both prognostic and predictive value in ATC patients. Additional studies are warranted to further elucidate the prognostic and predictive significance of MMR status in ATC.
Footnotes
Author Disclosure Statement
J.L. receives research support to the institution from Novartis, Bayer, and BMS, and consulting honoraria from Bayer, Genentech, and Ignyta. No competing financial interests exist for the remaining authors.