
Abstract
Graves' disease (GD) and Graves' orbitopathy are associated with stimulating thyrotropin receptor (TSHR) autoantibodies and autoreactive T cells. Recent in vitro studies suggested that sphingosine-1-phosphate (S1P) signaling is involved in the pathogenesis of orbitopathy. In this study, we explored the immune modulatory potential of S1P receptor antagonist fingolimod in a murine model for GD. Fingolimod was orally administered preventively during disease onset or therapeutically after disease onset. Administration of fingolimod during disease onset completely prevented the formation of TSHR-stimulating autoantibodies. Intervention after disease onset rarely reduced TSHR-stimulating autoantibodies and blocking autoantibodies were induced in some animals. Consequently, autoimmune hyperthyroidism characterized by elevated serum thyroxin levels, hyperplastic thyroid morphology accompanied by T cell infiltration, weight gain, enhanced body temperature, and tachycardia did not manifest preventively and showed milder manifestation in therapeutically treated animals. Importantly, examination of orbital tissue showed significant amelioration of orbitopathy manifestations through reduction of T cell infiltration, adipogenesis, and hyaluronan deposition. Autoimmune hyperthyroidism and orbitopathy were accompanied by changes in peripheral and splenic T cell proportions with high CD3+, CD4+, and CD8+ T cells. Activated T cells CD4+CD25+ were elevated whereas regulatory T cells CD4+Foxp3+ cells remained unchanged in spleens. Fingolimod decreased elevated T cell levels and increased CD4+CD25+Foxp3+ regulatory T cell populations. Analysis of total disease outcome revealed that treatment during disease onset protected animals against autoimmune hyperthyroidism and orbitopathy. Of note, therapeutic intervention after disease onset suppressed disease in half of the animals and in the other half disease remained at mild stages. The results of this study support a clinical trial to investigate the immunologic and clinical benefits of early treatment with S1P-based drugs in GD.
Introduction
Graves' disease (GD) is an autoimmune disorder caused mainly by autoantibodies against the thyrotropin receptor (TSHR) that bind to and chronically stimulate the receptor, leading to hyperthyroidism and thyroid hyperplasia (1 –3). Up to 30–50% of GD patients suffer from an auto-inflammatory eye disease termed Graves' orbitopathy (GO), but subclinical orbital involvement is found in all GD patients using imaging techniques (4 –6). GO is characterized by orbital inflammation, expansion of adipose tissue, and muscle fibrosis, resulting in variable symptoms of edema, protrusion of the eyeballs (proptosis), and muscle dysfunction (squint) (1).
Although overt manifestation of GO in GD patients is correlated with elevated levels of serum TSHR autoantibodies and severe hyperthyroidism, the disease has a multifactorial etiology where no single factor predicts the clinical outcome (7,8). Although hyperthyroidism of GD patients can be normalized by anti-thyroid drugs, limited therapeutic options are available to specifically treat GO and novel strategies are needed for early treatment of GD patients to prevent the development of orbitopathy.
Recent in vitro studies showed that sphingosine-1-phosphate (S1P) and receptor signaling is actively involved in the inflammation and remodeling processes in GO (9 –11). Moreover, our previous study revealed involvement of S1P in T cell attraction toward orbital fibroblasts as a disease-promoting mechanism (12). Collectively, these studies suggest targeting S1P signaling as a potential approach for controlling immune cell function as well as inflammatory and remodeling processes in the orbital tissue.
Among several S1P receptor modulators, fingolimod (FTY720) is already approved for oral immunotherapy of multiple sclerosis to decrease relapse rate and disability progression (13). As a functional S1P1 receptor antagonist, fingolimod prevents T cells from migrating and invading inflammatory sites, thereby reducing neuro-inflammation (14). Moreover, fingolimod has also been demonstrated to be effective in suppressing other autoimmune diseases, including thyroid or eye conditions in experimental and clinical settings (15 –17).
In this study, we investigated for the first time the efficacy of fingolimod in a preclinical mouse model for autoimmune hyperthyroidism and associated GO. In the model, autoimmune hyperthyroid animals showed TSHR-specific T cell responses and developed heterogeneity regarding GO with different degrees of orbital muscle inflammation and extension of adipose tissue, similar to that observed in patients (18 –20). In this study, we demonstrate that fingolimod suppresses the development of TSHR-stimulating autoantibodies and changes the proportions of T cell populations. Thus, fingolimod administered during disease onset prevented autoimmune hyperthyroidism and orbitopathy in animals, whereas treatment after onset of disease successfully suppressed progression of disease severity. The study suggests that the use of S1P receptor modulators may be a potential therapeutic strategy to modulate GD and GO.
Materials and Methods
Animals
Female BALB/c OlaHsd (H2d) inbred mice were purchased from a commercial supplier (Envigo GmbH, Netherlands) and housed in single ventilated cages in temperature- (23°C ± 1°C) and light-controlled (inverse 12:12 light-dark cycle) specific pathogen-free conditions. Food pellets and drinking water were provided ad libitum. All animal procedures were reviewed and approved by the North Rhine Westphalian State Agency for Nature, Environment and Consumer Protection (LANUV), Germany. We confirm that all experiments were performed in accordance to relevant guidelines and regulations.
Immunization procedure
Immunization of mice started at the age of 6 weeks, and they were sacrificed at the age of 21 weeks. For immunization, the eukaryotic expression plasmid pTriEx1.1Neo-human (h)TSHR A-subunit (aa1–289, also known as TSHT298) and the control pTri1Ex1.1Neo-β-Gal plasmid were used and administered as previously described (19,20). Briefly, 50 μg (1 mg/mL) of plasmid was injected into each biceps femoris muscle of isoflurane-sedated mice followed by electroporation. Mice were immunized four times in total with three-week intervals. Eight mice were treated with the control β-Gal (β-galactosidase) plasmid and 24 mice with the hTSHR A-subunit plasmid. All mice were monitored daily for general condition, and weight changes and body temperatures were determined.
Fingolimod treatment of TSHR-immunized mice
Mice immunized with TSHR A-subunit encoding plasmid (n = 24) were divided randomly into three groups (each n = 8) (Fig. 1). Two groups received fingolimod (FTY720; Cayman Chemical, Ann Arbor) through the drinking water (0.5 mg/kg body weight per mouse/day) either from the time point of the first immunization onward (TSHR +prev; treatment for 15 weeks) or from the time point of the last immunization onward (TSHR +ther; treatment for 6 weeks) until termination of the experiment 6 weeks after the last immunization, respectively.

Experimental design of fingolimod treatment in a mouse model for Graves' disease. To induce autoimmune hyperthyroidism and associated GO in the model, female BALB/c mice at the age of six weeks were immunized with TSHR A-subunit encoding plasmid four times at a three-week interval. TSHR-immunized mice were either treated with fingolimod from the first immunization onward to prevent disease during disease onset (TSHR +prev) or treated in a therapeutic manner starting with the last immunization when disease was already ongoing (TSHR +ther). Another group of TSHR-immunized mice remained untreated (TSHR). Female BALB/c mice immunized with β-Gal encoding control plasmid served as healthy control mice (β-Gal). The experiment ended six weeks after the last immunization. GO, Graves' orbitopathy; TSHR, thyrotropin receptor.
The untreated TSHR-immunized group (n = 8), as well as the β-Gal mice (n = 8), received the same volume vehicle dimethyl sulfoxide in drinking water as the fingolimod-treated mice from the time point of the first immunization onward (15 weeks). Water uptake was monitored daily, and water was changed twice weekly. Oral fingolimod treatment was safe and well tolerated in all mouse groups. Three TSHR A-subunit immunized mice died for unknown reasons.
Serological analyses
Total anti-human (h)TSHR antibodies, subtypes stimulating or blocking anti-mouse (m)TSHR antibodies, and total T4 were analyzed in sera as previously described (18 –20). Briefly, for anti-hTSHR antibodies 25 μL serum combined with 75 μL human control serum were measured by using a commercial TRAK kit following the manufacturer's instructions (ThermoFisher, BRAHMS, Germany).
Stably transfected mouse TSHR-CHO cells, kindly provided by Sandra McLachlan and Basil Rapoport, were used to perform a bioassay to measure stimulating or blocking activity of sera, as described elsewhere (21). The stimulating activity of antibodies is directly correlated with the cAMP production of the cells, which was measured by enzyme-linked immunosorbent assay (ELISA; Enzo, Farmingdale). Relative blocking activity was determined by measuring inhibition of cAMP production stimulated by a suboptimal dose of bTSH. Total T4 concentration was determined via ELISA (DRG, Springfield).
Echocardiography
Cardiac function was characterized by using murine transthoracic echocardiography (Vevo 2100; VisualSonics) as described earlier (22,23). In brief, three to four animals per group were randomly selected and sedated with isoflurane. Body temperature and respiratory rate were monitored and maintained stably. Left ventricular dimensions and systolic function were assessed in B-mode and M-mode. Cardiac output was calculated as the product of heart rate and stroke volume.
Histopathology and immunohistochemistry of thyroid and orbital tissues
Formalin-fixed and paraffin-embedded thyroids were cut (1 μm) and hematoxylin and eosin (H&E) stained. Thyroid morphology was analyzed objectively and blindly as well as indexed as normal, heterogeneous, or hyperplastic in comparison to the control mouse thyroid glands.
Formalin-fixed and paraffin-embedded orbits were cut (1 μm), and consecutive middle sections were used for histological analyses. Slices were stained with H&E and areas of nerve, fat, and muscle tissues were measured by using ImageJ as described earlier (19). We determined fat composition of retro-bulbar fat tissue based on characteristic differences in white and brown adipose tissue (BAT) morphologies. Since BAT is morphologically characterized by a much smaller vacuole size, both adipose tissue types can be clearly discriminated in H&E stained tissues. Fat composition was determined by measuring the areas of total fat and BAT in orbital sections. Percentages of BAT were calculated based on total orbital fat area. For visualization, BAT was stained specifically for UCP-1 (uncoupling protein-1, rabbit polyclonal IgG, dilution 1:1000, #UCP11-A; Alpha Diagnostics) as described earlier (19,24).
Further, orbital and thyroid sections were stained for CD3 (rabbit polyclonal IgG, dilution 1:25, # A0452; Dako) and with horseradish peroxidase (HRP)-conjugated-Polymer-Kit following the manual of the manufacturer (Zytomed Systems). Positive cells were counted in the orbital and thyroid sections and normalized to the area. Hyaluronic acid was detected in representative middle sections of the orbital tissue by using hyaluronic acid binding protein (HABP, dilution 1:250, # 385911; Merck). HABP binding was detected by using Avidin-Biotin Complex HRP kit (Vector Laboratories). Intensity of HRP-conjugated DAB staining was measured via ImageJ software and normalized to the area. Images were generated by using an Olympus BX51 microscope and Zeiss AxioObserver.
Flow cytometry of blood and splenic lymphocytes
Blood samples were incubated with erythrocyte lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA) for 2 minutes at room temperature and washed with phosphate-buffered saline (PBS) supplemented with 2% fetal calf serum and 2 mM EDTA. For the generation of single-cell suspensions, spleens were rinsed with erythrocyte lysis buffer. After sieving through a cell strainer, cells were washed with PBS supplemented with 2% fetal calf serum and 2 mM EDTA and resuspended in an appropriate volume.
Anti-CD3, Anti-CD4, Anti-8, Anti-CD19, Anti-CD25 (all BD Biosciences, Heidelberg, Germany), and Anti-Foxp3 (all eBioscience, Frankfurt, Germany) were used as fluorescein isothiocyanate, pacific blue, phycoerythrin, BD Horizon V450, allophycocyanin, AlexaFlour647, PE-cyanin 7, or peridinin-chlorophyll (all BD Biosciences) as protein conjugates. Dead cells were identified by staining with the fixable viability dye eFlour 780 (eBioscience). Intracellular staining for Foxp3 was performed with the Foxp3 staining kit (eBioscience) according to the manufacturer's recommendations. Flow cytometric expression analyses were performed with an LSR II instrument by using DIVA software (BD Biosciences) as described before (25,26).
Statistics
Statistical analyses were performed by using Prism 7 (GraphPad Software) and carried out with one-way ANOVA with Bonferroni post hoc test for multiple comparisons between mouse groups. Data are presented as mean ± standard error of the mean. Multiplicity adjusted p-values are marked as follows: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. Changes between mouse groups with p-values >0.05 are regarded as not statistically significant and are not marked in the graphs. In addition, the upper 99% confidence interval of the control β-Gal group was defined as threshold for positivity of individual mice and is indicated by a dotted line when appropriate.
The standard score (Z-score) was used to compare results from different mouse groups normalized to the mean value of the total mouse population (reference population). The Z-score values (arbritary units) represent the values of standard deviation from the mean value of the reference population. For objective grading of disease severity, Z-score values were chosen as thresholds as indicated.
Results
We investigated fingolimod as a potential treatment option in a mouse model for GD and associated orbitopathy (TSHR-immunized mice). To prove whether fingolimod is able to either prevent or improve disease, fingolimod was administered orally during disease onset (preventive setting; TSHR +prev) or after disease onset (therapeutic intervention; TSHR +ther) (Fig. 1). Mice immunized with β-galactosidase served as control mice (β-Gal).
Formation of TSHR-stimulating autoantibodies is prevented by fingolimod
Efficacy of fingolimod treatment on (auto)-immunity was determined by analysis of TSHR antibodies and their subtypes stimulating or blocking autoantibodies in mice sera (Fig. 2). Immunization of mice with the hTSHR A-subunit induced hTSHR antibody titer in sera of all TSHR-immunized mice to different degrees in individual animals (p ≤ 0.0001). Fingolimod applied to TSHR-immunized mice in a preventive manner strongly prevented the formation of hTSHR antibodies (p ≤ 0.01), whereas fingolimod did not significantly affect antibody formation in therapeutically treated mice compared with untreated TSHR-immunized mice (p > 0.05) (Fig. 2A).
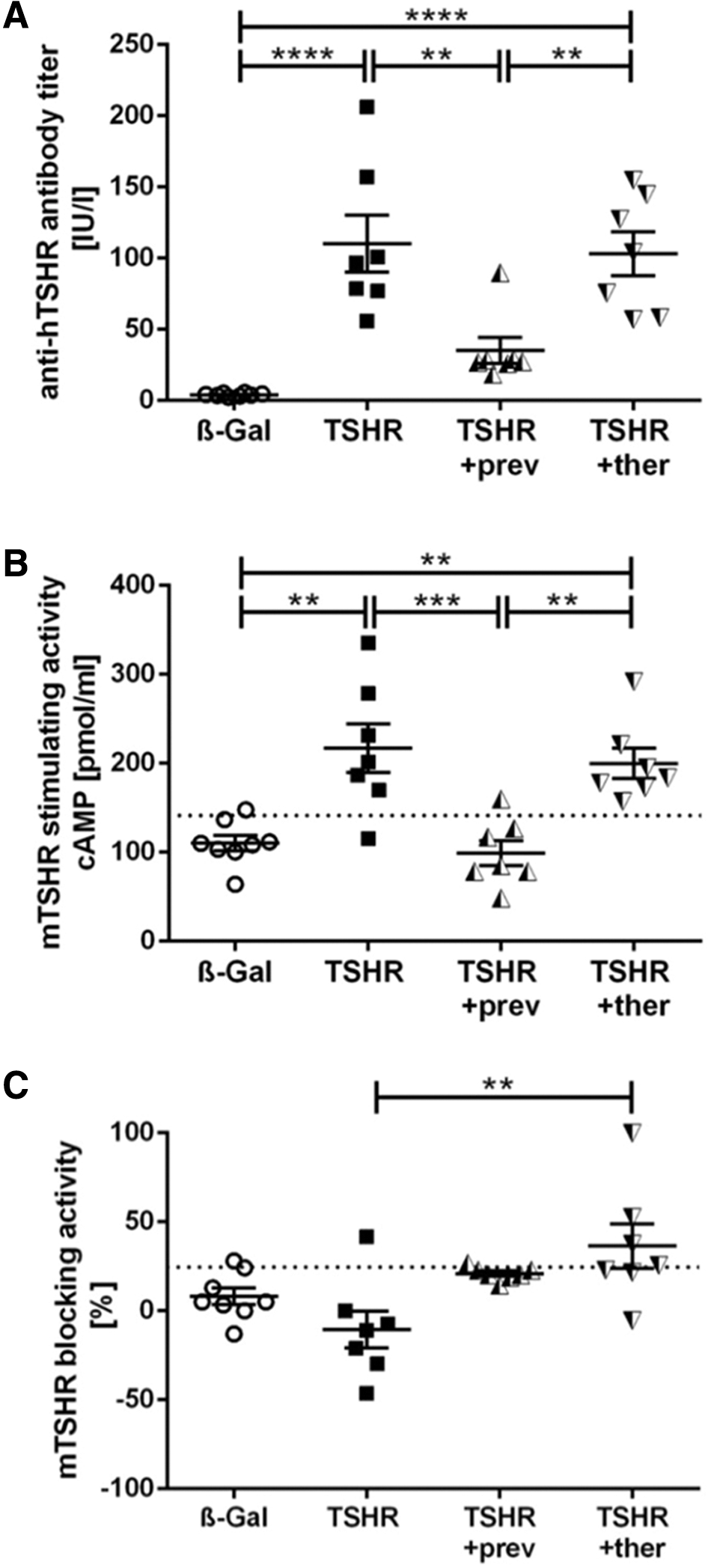
Effect of fingolimod treatment on formation of auto-antibodies. Detection of anti-TSHR antibodies in serum samples obtained from mice from each group β-Gal, TSHR, TSHR +prev, or TSHR +ther. (
Analysis of mouse TSHR-stimulating activity revealed an induction in 6 out of 7 animals in the TSHR-immunized mouse group (p ≤ 0.01), whereas preventive fingolimod treatment completely blocked the formation of stimulating autoantibodies in TSHR-immunized mice (p ≤ 0.001) (Fig. 2B). Therapeutic fingolimod treatment of TSHR-immunized mice did not significantly decrease stimulating autoantibodies (p > 0.05) (Fig. 2B). However, relative blocking activity of autoantibodies was increased in therapeutically treated mice when compared with the untreated TSHR mouse group (p ≤ 0.01) (Fig. 2C).
Fingolimod reduces or even inhibits autoimmune hyperthyroidism
We investigated the effect of fingolimod treatment on hyperthyroidism by measuring thyroid hormone T4 values in the sera and by evaluating thyroid histopathology (Fig. 3). Total T4 values were elevated in 6 out of 7 TSHR-immunized mice as a direct consequence of TSHR-stimulating activity of autoantibodies (p ≤ 0.01). In contrast, T4 values of preventively treated TSHR-immunized mice were not significantly increased compared with T4 values of the control β-Gal immunized group (p > 0.05) (Fig. 3A). However, T4 values of therapeutically treated TSHR-immunized mice remained elevated in 4 out of 7 mice whereas 3 out of 7 mice showed normal T4 values (p ≤ 0.05) (Fig. 3A).
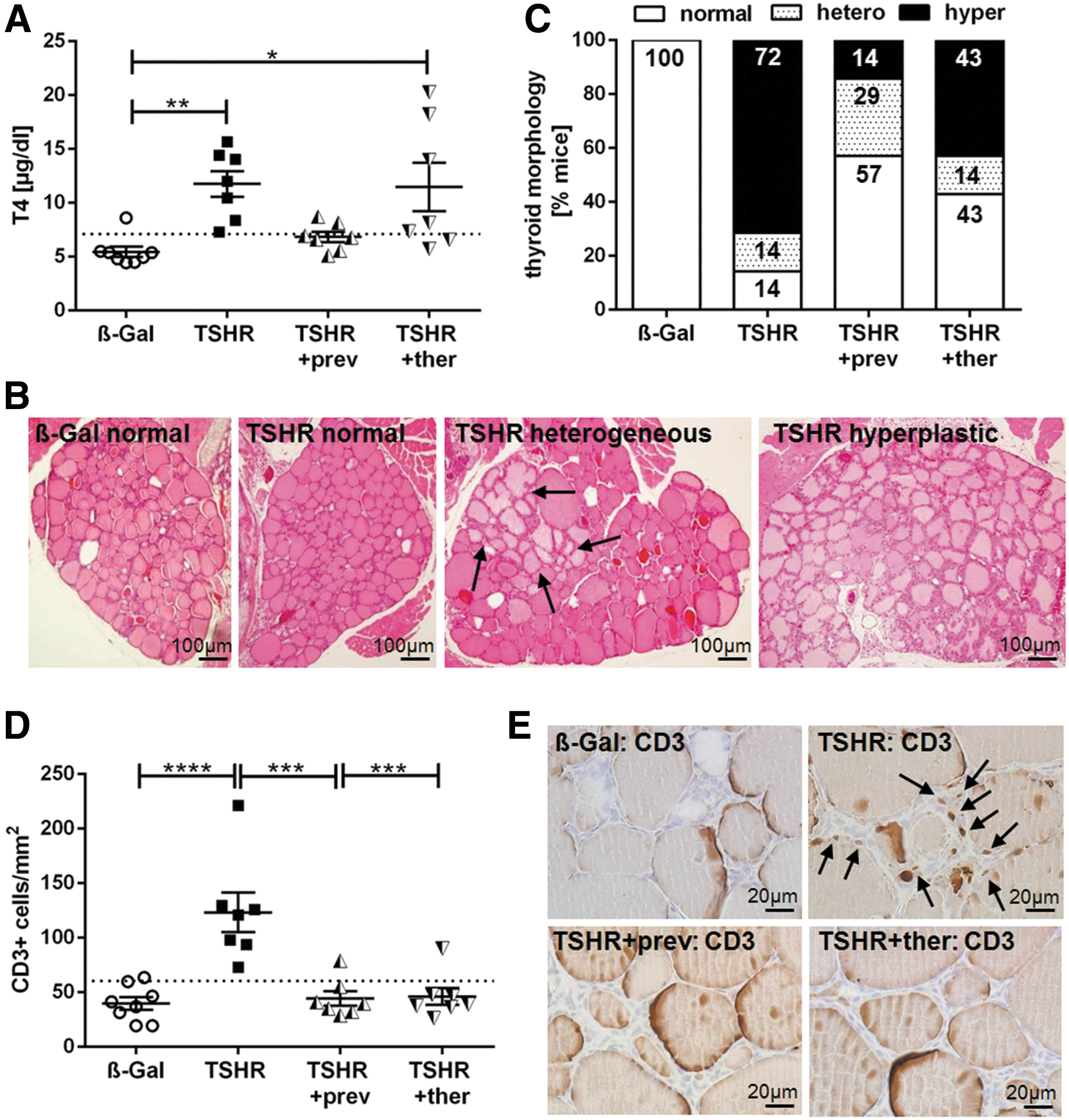
Improvement of thyroid dysfunction by fingolimod treatment. (
Thyroid sections were stained with H&E to seek for morphological changes, indicating dysfunction of the gland (Fig. 3B, C). Thyroids were categorized into normal, heterogeneous, and hyperplastic morphology. Hyperplastic morphology was characterized by cuboid cylindrical follicular cells with thick epithelium and a small amount of colloid, indicating follicular hyperactivity. Some of the glands showed heterogeneous morphology with a mixture of normally appearing zones and areas of hyperplastic follicles (Fig. 3B, arrows).
A morphology characteristic for hyperthyroidism was found in 72% of TSHR-immunized mice whereas only 14% of preventively and 43% of therapeutically treated mice remained morphologically hyperthyroid (Fig. 3C). Conversely, 57% of mice with preventive treatment and 43% of mice with therapeutic treatment showed a normal thyroid morphology, but only 14% of untreated mice showed a normal histology. The number of animals with a heterogeneous thyroid morphology also increased in the preventively treated group with 29% compared with untreated 14% in the TSHR-immunized and 14% in the TSHR +ther mice group (Fig. 3C).
Further, immunohistological staining of thyroid sections revealed extensive T cell infiltration into the interfollicular connective tissue in all TSHR-immunized mice (p ≤ 0.0001), which was prevented in almost all TSHR-immunized mice treated preventively or therapeutically with fingolimod (p ≤ 0.001; p > 0.05 compared with control β-Gal mice) (Fig. 3D, E).
Pathophysiological in vivo parameters are ameliorated by fingolimod
In addition, different physiological parameters were analyzed in living animals as an indication of hyperthyroidism. Body temperature was significantly increased in all TSHR-immunized mice compared with control β-Gal-immunized mice (p ≤ 0.001). Both fingolimod treatment protocols of the TSHR-immunized mice significantly diminished body temperature (p ≤ 0.0001), even though two of the therapeutically treated mice were found to have a decreased temperature (Fig. 4A). Some of the TSHR-immunized mice (4/8) tended to show a higher weight gain than control β-Gal mice, whereas preventively treated mice showed reduced weight gain (p ≤ 0.01). However, half of the therapeutically treated mice showed increased weight gain (4/8) (Fig. 4B).

Physiological in vivo parameters improved in response to fingolimod treatment. Six weeks after the last immunization, different physiological parameters have been measured in living mice, including (
Further, an increased heart rate occurred in TSHR-immunized animals (p ≤ 0.05), which could not be observed in the two treated groups (p ≤ 0.05) (Fig. 4C). Increased heart rate resulted in enhanced cardiac output in the untreated TSHR group (p ≤ 0.01), whereas cardiac output was not augmented in TSHR-immunized mice under preventive or therapeutic fingolimod treatment (p ≤ 0.01, p ≤ 0.001, p > 0.05 compared with β-Gal mice) (Fig. 4D). The normalized heart weight was moderately increased in TSHR-immunized mice and decreased with fingolimod treatment (Supplementary Fig. S1A).
GO is improved by fingolimod treatment
To determine to which degree fingolimod was able to reduce orbitopathy, consecutive sections of orbits were stained for T cells, BAT, and hyaluronan deposition (Fig. 5). Immunohistochemical CD3 staining revealed T cell infiltration into the orbital tissue in 6 out of 7 TSHR-immunized mice (p ≤ 0.001), which was entirely abolished by fingolimod treatment (p ≤ 0.001) (Fig. 5A, D upper panel, arrows).
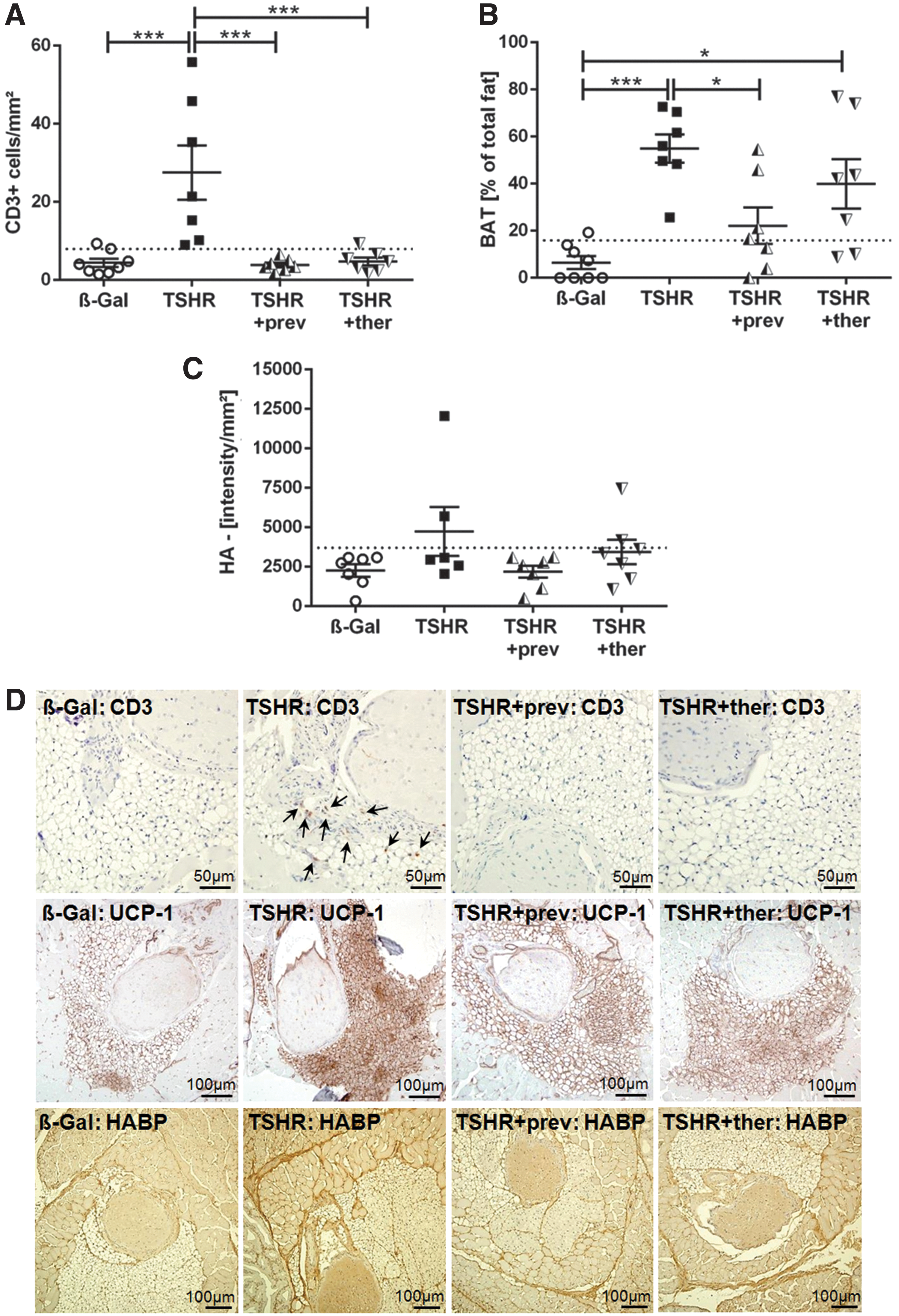
Orbital T cell infiltration and tissue remodeling improved on fingolimod treatment. The orbitae of all mice of each group were fixed and paraffin embedded; consecutive sections of the middle orbital area were subjected to different staining procedures. (
Earlier, we have shown that the mouse orbit contains a significant portion of TSHR-positive BAT that is enlarged in the mouse model (19,27). In agreement with our earlier studies, an increased percentage of BAT area was found in the orbital tissue of TSHR-immunized mice compared with control β-Gal-immunized mice (p ≤ 0.001) (Fig. 5B, D middle panel). In contrast, total orbital fat area was unaffected (data not shown). The group treated preventively with fingolimod showed a reduction of the BAT area in 4 out of 7 animals (p ≤ 0.05). The therapeutically treated mouse group still showed an increase in the BAT area, with 2 out of 7 animals displaying a normal percentage of BAT (p ≤ 0.05) (Fig. 5B). Interestingly, 4 out of 7 TSHR +ther mice that displayed the highest BAT enlargement showed also the highest T4 level (Fig. 3A).
Hyaluronic acid deposition was highly increased in orbital sections of some (2/6) TSHR-immunized mice (Fig. 5C, lower panel). In contrast, none of the preventive treated TSHR-immunized mice developed abnormal hyaluronan deposition. However, the orbital tissue of one TSHR +ther mouse also showed elevated hyaluronan amounts (Fig. 5C).
Fingolimod suppresses blood CD3+CD4+ and CD8+ T cells
To determine the impact of fingolimod treatment on lymphocyte distribution, whole-blood samples were analyzed by using flow cytometry after termination of the experiment (Fig. 6). Blood lymphocytes were moderately affected by fingolimod treatment. Four out of seven mice of each fingolimod treated group showed <0.5% lymphocytes and it is likely that this lymphopenia developed secondary to fingolimod treatment, a well-established adverse effect (Fig. 6A). The percentage of CD19+ B cells appeared to be unchanged in untreated or treated TSHR-immunized mice compared with β-Gal mice (Fig. 6B). In contrast, the percentage of CD3+ T cells was upregulated in the TSHR-immunized mouse group (p ≤ 0.001) and dramatically decreased in both fingolimod-treated groups (p ≤ 0.0001) (Fig. 6C).
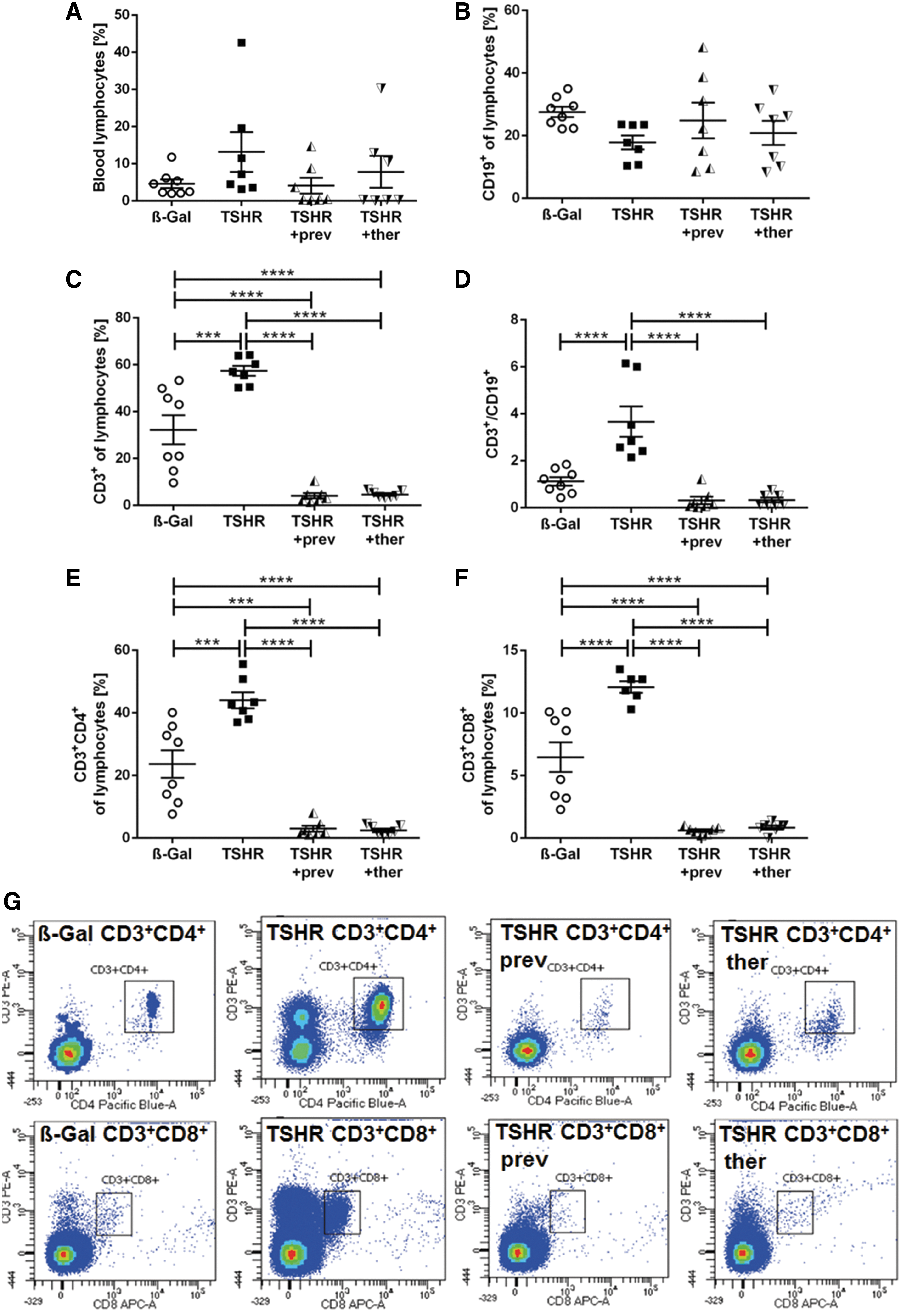
Modification of peripheral lymphocyte subsets by fingolimod. The lymphocyte composition of blood samples from β-Gal, TSHR, TSHR +prev, and TSHT +ther mice were analyzed by flow cytometry. (
The ratio of CD3+ T cells and CD19+ B cells was increased in TSHR-immunized mice (p ≤ 0.0001), and both fingolimod-treated groups showed levels similar to the control β-Gal group (Fig. 6D). Analysis of CD3+CD4+ and CD3+CD8+ T cell subtypes revealed that both subtypes were increased in TSHR-immunized mice and decreased with the two fingolimod protocols to a similar degree (p ≤ 0.001; p ≤ 0.0001, respectively) (Fig. 6E). The gating strategy is shown in Figure 6G.
Regulatory T cell subsets in spleen are differently affected by fingolimod
We further analyzed the effect of fingolimod treatment on splenocytes via flow cytometry (Fig. 7). Total splenic lymphocytes remained unchanged in TSHR-immunized mice and in the fingolimod-treated groups compared with β-Gal mice (Fig. 7A). CD4+ T cells were elevated in untreated TSHR-immunized mice but dramatically decreased in both fingolimod-treated groups, even below the levels found in controls (p ≤ 0.001; p ≤ 0.0001, respectively) (Fig. 7B). Similar results were obtained for CD8+ T cells, where preventively treated mice showed a stronger decline than the therapeutically treated group (p ≤ 0.0001; p ≤ 0.01) (Fig. 7C). Activated CD4+CD25+ T cells were elevated in TSHR-immunized mice (p ≤ 0.01), and fingolimod led to a decrease in both treatment groups (p ≤ 0.0001, p ≤ 0.001, respectively); preventively treated mice showed levels that were significantly below the ones found in controls (p ≤ 0.0001) (Fig. 7D). Regulatory CD4+Foxp3+ T cells were significantly diminished in mice preventively treated with fingolimod compared with control β-Gal or the TSHR-immunized mouse group (p ≤ 0.01; p ≤ 0.001) (Fig. 7E).
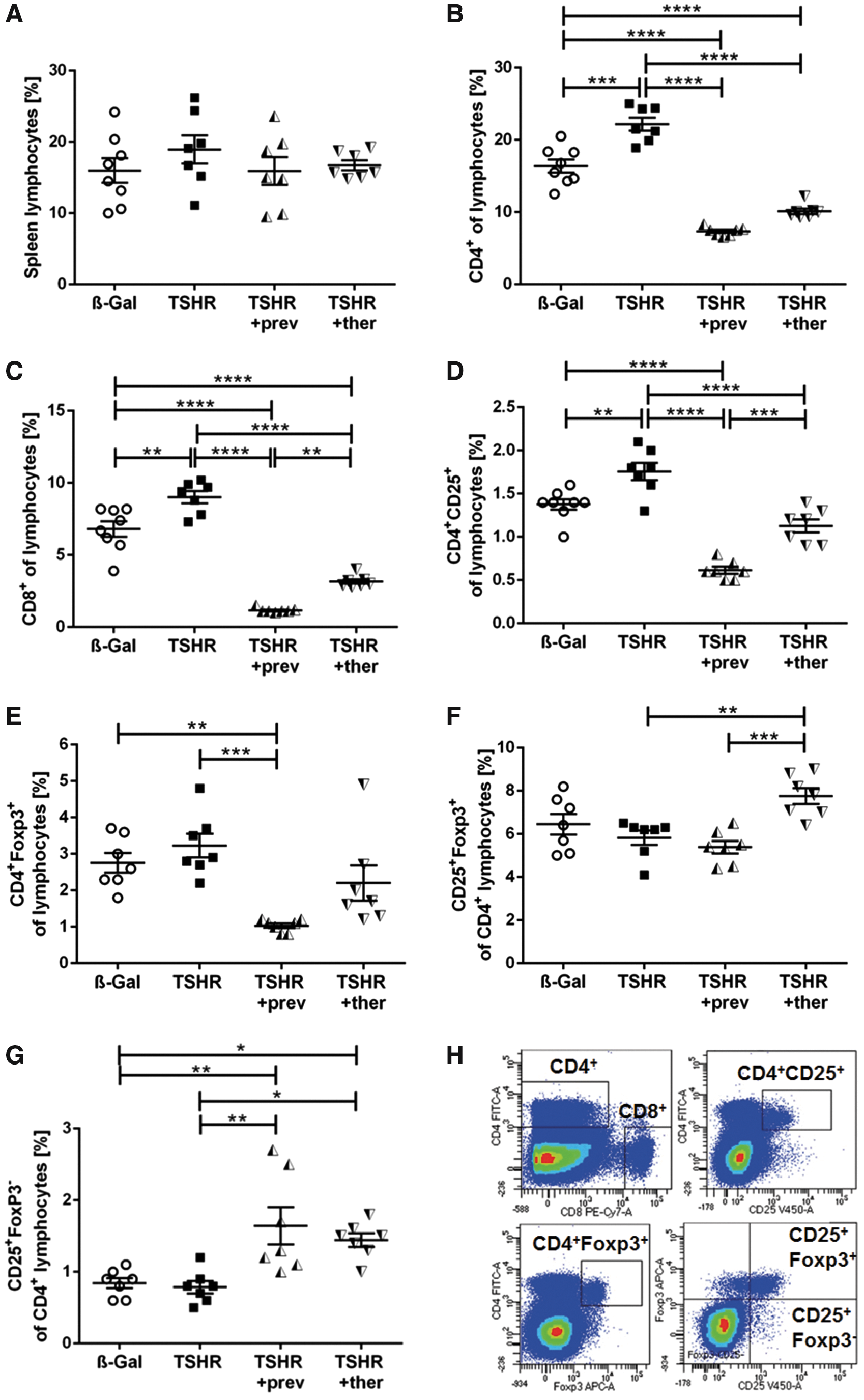
Splenic T cell subsets are affected by TSHR A-subunit immunization and fingolimod treatment. Spleens of the mice groups were homogenized and analyzed for T cell subsets by flow cytometry. (
TSHR-immunized mice that received fingolimod therapeutically showed significantly higher levels of CD4+CD25+FoxP3+ Treg subtypes in comparison to untreated and preventively fingolimod-treated mice (p ≤ 0.01; p ≤ 0.001) (Fig. 7F). The CD4+CD25+FoxP3− T cell subtype did not change in TSHR-immunized mice but it was enhanced in both fingolimod-treated groups (p ≤ 0.05; p ≤ 0.01, respectively) (Fig. 7G). However, CD4+CD25−FoxP3+ or CD4+CD25−FoxP3− subtypes were affected neither by immunization nor by fingolimod treatment (data not shown). The gating strategy is depicted in Figure 7H. The normalized spleen weight was moderately increased in TSHR-immunized mice and attenuated by fingolimod treatment (Supplementary Fig. S1B).
Fingolimod either inhibits or improves disease outcomes
Individual mice developed several signs of autoimmune hyperthyroidism or orbitopathy to different degrees. To compare total disease outcome between the mouse groups, we combined and normalized the different parameters for each mouse by the Z-score method described earlier (18,19). For the evaluation of total autoimmune hyperthyroidism, the data sets of five parameters (stimulating anti-mTSHR antibodies [TSAbs], T4 values, CD3+ T cells, temperature, and weight changes; Figs. 2 –4) were combined. For the evaluation of GO, the data sets of three parameters (CD3+ T cells, BAT and HA deposition; Fig. 5) were included.
As shown in Figure 8, the untreated TSHR mouse group (Z-score >0) can be clearly separated from the β-Gal mouse group (healthy control group; Z-score <0) with regard to total autoimmune hyperthyroidism (p ≤ 0.0001) and total GO (p ≤ 0.001) (Fig. 8A, B). Thus, TSHR-immunized mice manifested significant chronic disease during the experimental course (termed clinical disease). In contrast, treatment of TSHR-immunized mice with fingolimod resulted in a significant reduction of total autoimmune hyperthyroidism as well as total GO (Z-score <0), similar to the β-Gal control group (termed subclinical disease) (Fig. 8A, B).
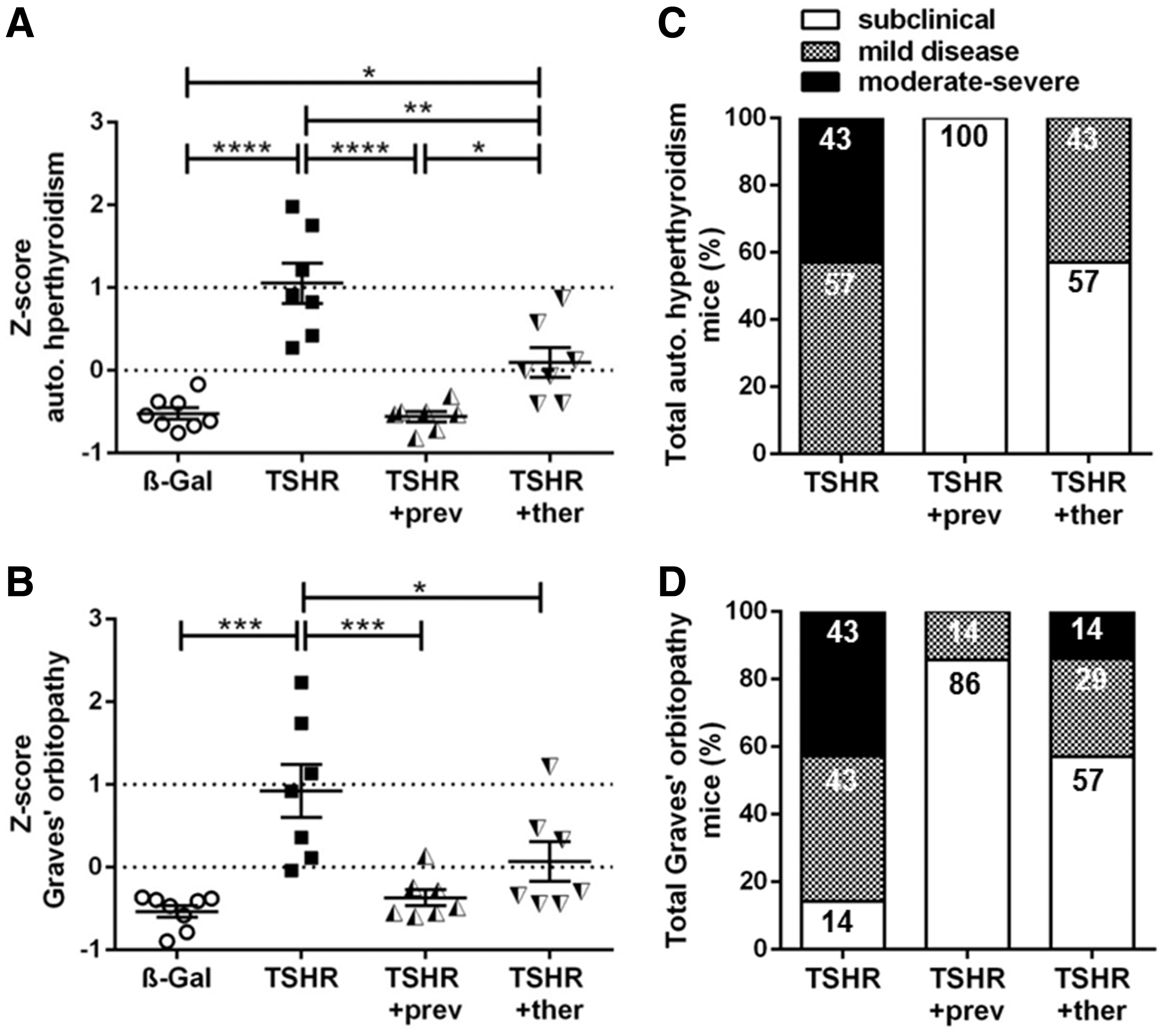
Total disease outcome of untreated and fingolimod-treated mice groups. Results of different experiments were combined and normalized by using the Z-score method. The Z-score represents the value of standard deviation from the mean value of the total mouse population and is given in arbitrary units. (
Finally, we categorized the disease outcome along Z-score values to evaluate the incidence and severity of disease. Analogous to the categories of GO severity in patients (28), we categorized clinical disease of mice into either mild or moderate-to-severe disease (Fig. 8C, D). For grading disease severity objectively in animals undergoing experimental GO, we selected a Z-score of 1 as a threshold to define mild disease (score 0 to 1), and a score >1 for moderate-to-severe disease.
These scoring categories are based on the observation that individual TSHR-immunized mice undergoing experimental GO develop a spectrum of abnormalities, resulting in a milder form or a more severe form of clinical disease. Immunized animals that had developed at least an increase of more than 50% in three out of five parameters describing autoimmune hyperthyroidism in the model, for example, a mouse that has shown a 50% increase in TSAbs, T4, and orbital CD3+ T cell infiltration (compared with a mean value of the β-Gal mouse group), were scored as “mild,” with Z-score values in the range of 0 > 1.
In contrast, a mouse that had shown more than a 100% increase in each of the three parameters was scored “moderate-to-severe” based on a Z-score >1. In terms of experimental GO, mice that had developed an increase of more than 50% (or 100%) in at least two out of three parameters describing GO in the model, for example, a mouse that had shown a 50% (or 100%) increase in CD3+ T cell infiltration and also in BAT enlargement (compared with a mean value of the β-Gal mouse group), were scored as “mild” based on a Z-score 0 > 1 or “moderate-to severe” if the Z-score was >1.
TSHR-immunized mice showing fewer changes than the mild category (Z-score values <0) were classified as subclinically diseased since these mice did not manifest significant autoimmune hyperthyroidism and/or orbitopathy, although these immunized mice might have developed TSHR antibodies (Fig. 2A). The percentages of TSHR-immunized mice in each category, that is, subclinical or clinical (mild or moderate-to-severe), are shown in Figure 8C and D. In addition, the number of mice categories are given in Supplementary Table S1.
Regarding autoimmune hyperthyroidism, all untreated TSHR-immunized mice developed clinical disease (p ≤ 0.0001), and 43% even developed moderate-to-severe disease (Fig. 8A, C). In contrast, 100% of preventively treated mice (p ≤ 0.0001), and 57% of the therapeutically treated mice did not manifest clinical autoimmune hyperthyroidism; whereas 43% of the TSHR +ther mice developed mild autoimmune hyperthyroidism (p ≤ 0.05) (Fig. 8A, C).
Regarding orbitopathy, 86% of TSHR-immunized mice developed either mild or moderate-to-severe disease and 14% remained subclinical (p ≤ 0.001) (Fig. 8B, D). In contrast, 86% of TSHR +prev mice, as well as 57% of TSHR +ther mice, showed no signs of orbitopathy compared with untreated TSHR-immunized mice (p ≤ 0.001; p ≤ 0.05). However, 14% of TSHR +prev mice and 29% of TSHR +ther mice were mildly affected, and 14% of TSHR +ther mice had persistent moderate-to-severe orbitopathy (Fig. 8B, D).
Discussion
In this study, we have examined the efficacy of fingolimod, a functional S1P receptor antagonist and a modulator of T cell circulation, on disease progression in a murine model for GD. We demonstrate that fingolimod inhibits development and progression of autoimmune hyperthyroidism and associated orbitopathy in animals immunized with the hTSHR A-subunit. Fingolimod administered during disease onset blunted production of autoantibodies, efficiently modulated T cell proportions, and protected thyroid and orbital tissue from T cell invasion, thereby preventing hyperthyroidism and orbitopathy. Moreover, we show that intervention with fingolimod after disease onset limits the severity of disease.
Modulation of TSHR autoimmunity by fingolimod
Several reports have suggested a relative imbalance between T effector cells and regulatory T cells that may account for the induction of thyroid autoimmunity and orbitopathy, even though the exact contribution of each T cell subset remains unclear (29 –34). Indeed, in our murine model, autoimmune hyperthyroidism and orbitopathy was accompanied by changes in peripheral and splenic T cell proportions, whereas B cells remained unaffected. Peripheral and splenic CD4+ and CD8+ T cells and splenic CD4+CD25+ activated T cells were elevated, indicating ongoing immune responses to the hTSHR A-subunit antigen. However, splenic CD4+Foxp3+ and CD4+CD25+Foxp3+ T cell levels were unchanged in this model. In contrast, in a GD mouse model induced by the adenovirus encoding hTSHR289, a decrease in the percentage of splenic CD4+CD25+Foxp3+ Tregs was found (35,36) and it was shown earlier that depletion of CD4+CD25+ T cells aggravates hyperthyroidism in the GD mouse model (37).
However, in the treatment study presented here, we confirm the well-known immunosuppressive function of fingolimod by demonstrating a clear reduction of elevated T cell subsets in blood and spleen, including CD3+, CD4+, and CD8+ lymphocytes. Further, we found that fingolimod in either setting reduced elevated CD4+CD25+-activated T cells and diminished CD4+Foxp3+ Treg levels, a finding that is in line with studies with fingolimod therapy in experimental autoimmune thyroiditis using nonobese diabetic (NOD) mice or in experimental autoimmune uveitis (16,17). Preventive treatment in our model led to a more prominent decrease in these T cell subsets, which is most likely explained by the longer duration of therapy in the preventive compared with therapeutic protocol (15 weeks vs. 6 weeks).
Of note, the therapeutic protocol led to an increase in the percentage of splenic CD4+CD25+Foxp3+ Tregs, which might reflect an earlier response to thyroid/orbital tissue inflammation and suggests that a mechanism to control autoimmunity has been initiated. Unexpectedly, in the preventive protocol used in our model, splenic CD4+CD25+Foxp3+ Tregs appeared unchanged. An explanation would be that Tregs increased and declined during the prolonged experimental course.
Further, the absence of autoantibodies and thyroidal/orbital inflammation might lead to a lack of induction of this Treg type. Interestingly, fingolimod in either setting led to an increase in the CD4+CD25+Foxp3− T cell subset, most likely indicating a decrease in FoxP3 expression in the CD4+CD25+-activated T cell population during immune suppression. Whether the observed changes in peripheral and splenic T cell proportions in response to fingolimod treatment represent their role in improving autoimmune hyperthyroidism and orbitopathy in the model remains to be proved by further studies addressing the functional activity of regulatory T cell subsets.
Another significant finding in our study is that the formation of TSHR-stimulating autoantibodies in preventively treated mice was blunted, most likely as a result of reduced peripheral CD4+ T cell levels and thus T cell helper function. Peripheral B cell levels were unchanged in response to fingolimod treatment, a finding that is in line with a recent study on an experimental autoimmune encephalomyelitis mouse model (38). However, the results of that study suggested a modulation of B cell receptor-mediated signaling, a mechanism that could also have contributed to a reduction of antibody formation or activity in our model. Consequently, autoimmune hyperthyroidism characterized by elevated T4 levels and a hyperplastic thyroid morphology accompanied by T cell infiltration was entirely inhibited by preventive fingolimod treatment.
In contrast, fingolimod therapy initiated after disease onset had a marginal effect on already present TSHR autoantibodies, even though it might have hindered a raising production of stimulating autoantibodies. Interestingly, fingolimod treatment induced TSHR-blocking autoantibodies in some of the animals. Blocking antibodies can inhibit cAMP-dependent pathways in target tissues and may contribute to a decrease in thyroxine levels or orbital adipogenesis (2). In our study, the mixture of stimulating and/or blocking autoantibody levels in therapeutically treated animals led to autoimmune hyperthyroidism in 50% of the animals; whereas thyroid function remained normal or normalized in the other 50%.
Fingolimod normalizes thyroid dysfunction and reduces orbitopathy
We also investigated the effect of fingolimod on physical parameters in TSHR-immunized mice. Untreated TSHR-immunized mice showed an increase in body temperature, weight gain, and tachycardia, indicating severe hyperthyroidism, findings that are in agreement with other studies (39,40). Preventive and therapeutic treatment with fingolimod significantly decreased tachycardia that had developed in untreated TSHR-immunized mice. Multiple sclerosis patients frequently develop a transient bradycardia as a direct effect of fingolimod therapy, which is observed one to two hours after treatment with an attenuation after six hours (41 –43). This phenomenon is triggered by the initial agonistic effect of fingolimod on the S1P1 receptor expressed on myocytes.
Our study suggests a normalization of cardiac function after long-term treatment over months as a consequence of the systemic immunosuppressive property of fingolimod rather than as a direct effect on myocytes. Similarly, Holthoff et al. showed that immunotherapy with cyclic peptides leads to a decrease of tachycardia, which was shown to occur consistently in the long-term Ad-TSHR289-immunized mouse model over three to nine months (40). However, in contrast to our results, heart rate was not normalized by the treatment with cyclic peptides (44,45). In addition, it was shown that hyperthyroidism in mice leads to an increase in body temperature and increased weight gain (39).
In our study, both increased parameters in untreated TSHR-immunized mice were normalized through preventive and therapeutic treatment with fingolimod. Both preventive and therapeutic fingolimod treatment led to an inhibition or reduction of hyperthyroidism, based on physiological parameters. Of note, none of the preventive or therapeutically treated animals developed hypothyroidism based on serum thyroxine levels, and thyroid morphology and in vivo results indicate that fingolimod neither directly alters thyroid function nor indirectly alters the hypothalamic-pituitary-thyroid axis, which would be advantageous compared with the currently used antithyroid drugs.
Importantly, fingolimod was able to reduce orbitopathy characterized by T cell infiltration, BAT enlargement, and hyaluronan deposition. CD3+ T cell infiltration of orbital tissue was completely blocked in all mice under fingolimod treatment, findings that are consistent with a study showing that fingolimod prevents lymphocyte migration to the eyes and decreased CD4+CD25+ as well as CD4+CD25+Foxp3+ T cell levels in the eyes of experimental mice with autoimmune uveitis (16).
Earlier, we have shown that orbital BAT is enlarged in response to TSHR- immunization in this mouse model (19). This is in line with studies showing that the formation and activity of BAT could be increased by activation of the TSHR and high thyroid hormone levels (46,47). In addition, BAT expresses high amounts of S1P1 receptor and is regulated by S1P (48), which implies that fingolimod, in addition to its immunomodulatory effects, can have a direct effect on orbital BAT development. Of note, fingolimod has shown anti-obesity property by inhibiting adipocyte differentiation and prolypolytic action in diet-induced obesity in mice (49).
In our study, we found that orbital BAT enlargement was reduced in 4 out of 7 preventively fingolimod-treated animals whereas 3 out of 7 preventively treated animals still developed enlarged BAT. These findings indicate that other mechanisms than TSHR activation, for example, high thyroid hormone levels and/or S1P1 receptor signaling may contribute to BAT enlargement in the model. Moreover, elevated orbital hyaluronan deposition observed only in a few TSHR-immunized mice was completely prohibited by preventive treatment and therefore might be fully dependent on the action of TSHR-stimulating autoantibodies. Since deposition of hyaluronan in the orbits will be likely more prominent in later phases of orbital disease, an extended study is needed to fully elucidate the impact of fingolimod on fibrosis, as shown in this model 15 weeks after the last immunization (20).
Efficacy of immunotherapy of GD/GO using fingolimod
To compare the efficacy of fingolimod in a preventive versus a therapeutic setting, we analyzed the outcomes of total autoimmune hyperthyroidism and orbitopathy. Intervention during disease onset was superior to intervention after disease onset in inhibition of autoimmune hyperthyroidism and orbitopathy. In both settings, fingolimod showed higher efficacy in the treatment of autoimmune hyperthyroidism than orbitopathy based on histopathological parameters included in this study. The high efficacy of fingolimod as a preventive treatment is attributed to the inhibitory effect against the development of TSHR-stimulating autoantibodies. A variety of studies have been performed to investigate the efficacy of preventive and therapeutic fingolimod treatment in different diseases.
Fingolimod was shown to reduce the formation of thyroglobulin antibodies in experimental Hashimoto's thyroiditis or acetylcholine receptor antibodies in experimental myasthenia gravis when administered during disease onset (15,50). In contrast, no change in either antibody titers or antigen-specific plasma cell populations and disease severity could be shown for myasthenia gravis when fingolimod was administrated after disease onset (51). Likewise, an NOD mouse model of diabetes mellitus showed complete suppression of disease development by preventive treatment with fingolimod. Therapeutic treatment with fingolimod resulted in prolongation of survival and protection of β cells against autoimmune destruction (52).
In contrast, numerous studies on experimental autoimmune encephalitis (EAE), an animal model for multiple sclerosis, revealed that fingolimod is effective both preventively and therapeutically by reducing the recruitment of autoaggressive T cells to disease-relevant tissues (53 –57). Interestingly, late-stage therapy with fingolimod resulted in a rescue by reversing neurologic deficits in EAE mice (58). In line with these studies, the results of our study also reveal a higher efficacy of preventive treatment, suggesting that treatment with fingolimod should be initiated as soon as the disease is diagnosed.
A different immunomodulatory approach to improve GD and GO outcomes in an adenoviral A-TSHR298 mouse model consists of the treatment with TSHR-derived cyclic peptides to induce T cell and/or B cell anergy (44). Therapeutic treatment with cyclic peptide 836 one week after the fourth immunization led to a reduction of T4 levels starting 15 weeks after therapy onset. Further, orbital fibrosis, tachycardia, and cardiac hypertrophy were improved. However, levels of stimulating TSHR antibodies were not affected by this therapy and T cells and/or B cells have not been investigated to prove immune modulation (43).
Recently, an attempt to control GD emerged, which made use of tolerogenic peptides for TSHR antigen-specific immunotherapy (59). These so-called apitopes (antigen-processing independent epitopes) mimic naturally processed CD4+ T cell epitopes to induce tolerance to a self-antigen without being processed by antigen-presenting cells. The study in HLA-DR3 transgenic mice showed that the apitope ATX-GD-59 was able to induce tolerance ex vivo in splenocytes and lymph nodes. Most importantly, prophylactic treatment of a GD mouse model immunized with an adenovirus encoding TSHR298 reduced anti-TSHR antibody levels but did not reduce serum T4 levels. However, orbitopathy was not investigated in the latter study (58).
New approaches to modulate the immune system in GO patients have been tested in therapeutic trials. Rituximab (RTX) used in open-label series has been associated with very encouraging responses in patients with active and moderate-to-severe GO depending on patient populations treated (60,61). RTX is a chimeric mouse-human monoclonal antibody targeting CD20 that results in a depletion of B cells in intermediate stages of maturation and short-lived plasma cells (62,63). Interestingly, RTX treatment also reduced peripheral IGF1R+ CD4+ and CD8+ T cells in GO patients (64). It might be considered as early treatment, especially in severe corticosteroid-resistant cases. As illustrated for fingolimod in this study, it is conceivable that early treatment with RTX may also be associated with higher efficacy (61,65).
In conclusion, the results of this study suggest that fingolimod is a good candidate for the early treatment of human GD and GO as a disease-modifying drug and should be investigated in clinical trials.
Footnotes
Acknowledgments
The authors gratefully acknowledge Drs. Sandra McLachlan and Basil Rapoport for generously providing stably transfected mouse TSHR-CHO cells. They are most grateful to Mareike Horstmann and Christoph Jesenek (University Hospital Essen) for excellent technical assistance. They also thank Alexandra Brenzel (Imaging Center Essen IMCES, University Duisburg-Essen) for technical assistance. The study was supported by funding by Deutsche Forschungsgemeinschaft DFG grant GRK 2089 (A.E., U.B.P., W.H.) and by IFORES research grant from the Medical Faculty, University Duisburg-Essen (L.M.).
Author Disclosure Statement
All authors disclose any commercial association that might create a conflict of interest in connection with this article.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1