
Abstract
Background:
Glycogen storage disease type Ia (GSD Ia), also known as von Gierke disease, is the most common glycogen storage disorder. It is caused by the deficiency of glucose-6-phosphatase, the enzyme that catalyzes the final step of gluconeogenesis and glycogenolysis. The accumulation of glucose-6-phosphate leads to increased glycogen and triglyceride levels in the liver. Patients with GSD Ia can develop steatohepatitis, cirrhosis, and increased risk for hepatocellular adenomas and carcinomas. We previously showed that animal models of GSD Ia had defective autophagy and dysfunctional mitochondria. In this study, we examined the effect of VK2809, a liver-specific thyroid hormone receptor β agonist, on hepatic steatosis, autophagy, and mitochondrial biogenesis in a mouse model of GSD Ia.
Methods:
G6pc −/−-deficient (GSD Ia) mice were treated with VK2809 or vehicle control by daily intraperitoneal injection for four days. The hepatic triglyceride and glycogen were determined by biochemical assays. Autophagy and mitochondrial biogenesis were measured by Western blotting for key autophagy and mitochondrial markers.
Results:
VK2809 treatment decreased hepatic mass and triglyceride content in GSD Ia mice. VK2809 stimulated hepatic autophagic flux as evidenced by increased microtubule-associated protein light chain 3-II (LC3B-II), decreased p62 protein levels, activation of AMP-activated protein kinase (AMPK), inhibition of the mammalian target of rapamycin (mTOR) signaling, enhancement of protein levels of ATG5-ATG12, and increased lysosomal protein expression. VK2809 also increased the expression of carnitine palmitoyltransferase 1a (CPT1α) and fibroblast growth factor 21 (FGF21), as well as mitochondrial biogenesis to promote mitochondrial β-oxidation.
Conclusions:
In summary, VK2809 treatment decreased hepatic triglyceride levels in GSD Ia mice through its simultaneous restoration of autophagy, mitochondrial biogenesis, and β-oxidation of fatty acids. Liver-specific thyromimetics represent a potential therapy for hepatosteatosis in GSD Ia as well as nonalcoholic fatty liver disease.
Introduction
Glycogen storage disease type Ia (GSD Ia), also known as von Gierke disease, is the most common glycogen storage disease. It is caused by loss-of-function mutations in the G6PC gene, resulting in decreased enzymatic activity of glucose-6-phosphatase (G6Pase) (1), which catalyzes the hydrolysis of glucose-6-phosphate (G6P) to free glucose, the common final step in both glycogenolysis and gluconeogenesis (2). G6Pase is primarily expressed in the liver, and to a lesser extent, in the kidney and intestines. Patients with GSD Ia can have severe hypoglycemia during fasting (3), increased serum levels of uric acid, lactic acid, triglycerides, and very-low-density lipoprotein (4), and long-term complications due to metabolic derangements in the affected organs (5). Disease management, consisting of dietary therapy with frequent feedings with complex carbohydrates, has enabled GSD Ia patients to survive into early adulthood; however, such measures do not prevent long-term complications, such as progressive liver failure and cirrhosis (6).
The liver is the major affected organ in GSD Ia. Hepatic deficiency of G6Pase leads to impaired gluconeogenesis and accumulation of G6P, which then leads to increased glycogen and fatty acid synthesis (7,8). Patients develop hepatomegaly due to the increases in glycogen and triglyceride content. GSD Ia patients develop hepatic steatosis at a young age (4) and later develop long-term complications, such as steatohepatitis and cirrhosis. Additionally, 70–80% of GSD Ia patients older than 25 years develop hepatocellular adenomas and have an increased risk for developing hepatocellular carcinoma (5).
Another liver derangement observed in GSD Ia is mitochondrial dysfunction, which is also thought to be involved in the pathogenesis of many other metabolic disorders, including nonalcoholic fatty liver disease (NAFLD) (9,10). Loss of G6Pase function leads to reduced mitochondrial content, mitochondrial respiration, altered metabolites of tricarboxylic acid cycle (TCA cycle) intermediates, and accumulation of triglycerides (9). These observations are in line with the previous findings that showed G6Pase-deficient hepatocytes have increased anaerobic metabolism (11,12). Thus, increasing mitochondrial biogenesis and removing damaged mitochondria may improve lipid metabolism and oxidative phosphorylation in GSD Ia. These changes, in turn, may resolve or delay the long-term hepatic complications in GSD Ia patients.
(Macro)autophagy is a lysosome-dependent degradation process. It involves the formation of double-membrane vesicles termed “autophagosomes,” the sequestration of cytosolic substrate within autophagosomes, and the subsequent fusion of autophagosomes and lysosomes to form autolysosomes. Engulfed macromolecules such as lipids and glycogen, as well as damaged proteins and organelles, are degraded to maintain cellular homeostasis (13 –15). Deficient autophagy has been implicated in the pathogenesis of various liver diseases associated with hepatosteatosis, including both GSD Ia and NAFLD (8,16,17). Of note, pharmacological restoration of autophagy increased lysosomal hydrolysis of triglycerides and reduced hepatosteatosis in animal models of GSD Ia and NAFLD and thus may represent a novel therapeutic strategy for treating these conditions (18,19).
Thyroid hormone (TH) regulates hepatic lipid metabolism and is able to reduce hepatosteatosis and hypercholesterolemia (20 –23). However, its adverse effects on the heart and bone are major concerns for clinical application (24). VK2809 (formerly known as MB07811) is a prodrug, which undergoes first-pass hepatic extraction and requires cytochrome P450 cleavage to generate the negatively charged thyroid hormone receptor β (THRβ) agonist, VK2809A (formerly known as MB07344) (25). It distributes poorly into most tissues and is rapidly eliminated in the bile (26). Those features make VK2809 a highly liver-specific thyromimetic since it did not demonstrate any adverse cardiac side effects (25). At the molecular level(s), TH increases hepatic autophagy, which is critical for induction of mitochondrial activity, mitochondrial biogenesis, and lipid oxidation (27 –31). We previously observed that supplemental levothyroxine therapy was able to decrease hepatosteatosis in diabetic patients (20). We thus hypothesized that TH and/or thyromimetics could be beneficial for GSD Ia and assessed the effects of VK2809 on hepatosteatosis, autophagy, and mitochondrial biogenesis in murine GSD Ia.
Materials and Methods
Animal models
Animal studies were approved by the Duke University Institutional Animal Care and Use Committee under the protocols A038-17-02. All animals received care according the criteria outlined in the National Institutes of Health (NIH) publication 86–23.
Heterozygous G6pc+/− mice were housed in the Duke Vivarium and bred to produce homozygous G6pc−/− offspring, which are referred to as GSD Ia mice in the remainder of the article. Genotype was confirmed by polymerase chain reaction analysis of tail DNA with primers within and flanking the neo gene insertion in the G6Pase gene as has been previously published (32). At day 5 after birth, wild-type (WT) or GSD Ia mice were intraperitoneally injected with VK2809 (10 mg/kg body weight [BW]) or vehicle (3% Pharmasolve, 20% polyethylene glycol 400, 8% ethanol, and 10% Solutol HS15 in water, pH ∼6–7) daily for four days. Supplemental glucose was administered twice daily to GSD Ia mice as 10% dextrose (10 mL/kg body weight) injected subcutaneously, up to day 8. At nine days of age, mice were euthanized, and tissues were collected for subsequent analysis. The groups analyzed were as follows: WT+Vehicle (n = 6), WT+VK2809 (n = 7), GSD Ia+Vehicle (n = 6), and GSD Ia+VK2809 (n = 7).
Glycogen content and triglyceride analyses
Triglyceride concentrations in the liver and serum were measured using Triglyceride Colorimetric Assay Kits (10010303; Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer's instructions. Glycogen content assays were performed on liver homogenates using previously described methods (33).
Western blotting
A 10 mg frozen liver sample was homogenized in 200 μL of cold radioimmunoprecipitation assay buffer (RIPA buffer) (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 1% sodium dodecyl sulfate [SDS], 0.5% sodium deoxycholate) supplemented with phosphatase inhibitor and protease inhibitor (Sigma Chemical Co., St. Louis, MO) using MagNA Lyser (Roche, Basel, Switzerland). Protein concentration was determined by BCA Assay Kit (Thermo Fisher Scientific, Waltham MA). Then, 40 μg of protein was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 6–16% gradient Tris-glycine gels and transferred to 0.45 mM polyvinylidene fluoride membrane (Millipore, Burlington, MA). Membranes were blocked in 5% milk/phosphate-buffered saline with Tween-20 (PBST), and antibodies were incubated overnight at 4°C in 1% bovine serum albumin/PBST. This was followed by incubating with the secondary antibody conjugated with horseradish peroxidase and visualized using the enhanced chemiluminescence system (GE Healthcare, Chicago, IL). The following antibodies were used to detect the target proteins: Cell Signaling Technology (Danvers, MA): LC3B (2775), p62 (5114), COXIV (4850), ATG5-ATG12 (2630), AMPK (5831), p-AMPK (Thr172) (2535), MTOR (2983), p-MTOR (Ser2448) (5536), 4EBP1 (9452), p-4EBP1 (Thr37/46) (2855), VDAC1 (4661), and FAS (3180); Santa Cruz Biotechnology (Dallas, TX): actin (sc-47778), cathepsin B (sc-13985), and cathepsin D (sc-6486); Abcam (Cambridge, UK): p-ATGL (Ser406) (ab135093), ATGL (ab99532), ERRα (07-662), and CPT1α (ab128568); Sigma Chemical Co.: PGC1α (SAB2500781); Thermo Fisher Scientific: LAMP2 (PA1-655). Densitometric analyses of Western blot images were performed by using the ImageJ software (NIH, Bethesda, MD). Phosphorylated protein was normalized to total protein, and others were normalized to actin.
Measurement of serum total thyroxine
Serum total thyroxine (T4) concentrations were measured by radioimmunoassay, as detailed previously (34).
Statistics
Statistical analysis was performed using GraphPad Prism 8. Statistical significance was determined by two-way analysis of variance followed by Fisher's least significant difference multiple comparisons test. The statistical significance of comparisons is indicated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Outliers were determined by GraphPad QuickCalcs: outlier calculator, and significant outliers (p < 0.05) were excluded from statistical analysis.
Results
VK2809 treatment reverses hepatomegaly and hepatosteatosis in GSD Ia
The phenotype in GSD Ia mice closely mimics GSD Ia in humans as these mice develop severe hypoglycemia, hepatomegaly due to the accumulation of glycogen and triglyceride, and low body weight shortly after birth (32). Moreover, only 15% of GSD Ia mice survive weaning by 21 days of life, despite injections with supplemental glucose (32). Accordingly, we initiated our studies when mice were 5 days old and gave daily intraperitoneal injections of VK2809 (10 mg/kg BW) or vehicle for four days before sacrificing the mice and collecting their livers and sera.
GSD Ia mice have significant hepatomegaly; however, VK2809 treatment markedly decreased the liver mass of GSD Ia mice to levels similar as those observed in WT mice (Fig. 1A). After normalization for body weight, the ratio of liver mass to body weight also showed a significant decrease in GSD Ia mice treated with VK2809 (Supplementary Fig. S1A). VK2809 slightly increased mortality of GSD Ia mice at day 9 (Supplementary Fig. S2). The cause of mortality remains unknown; however, these mice are fragile and develop hypoglycemia despite of glucose supplementation. Of note, we found that VK2809 treatment decreased the serum total T4 levels in both GSD Ia and WT mice (Supplementary Fig. S3); however, WT mice treated with VK2809 did not have any change in mortality suggesting that reduced endogenous T4 may not be contributory.
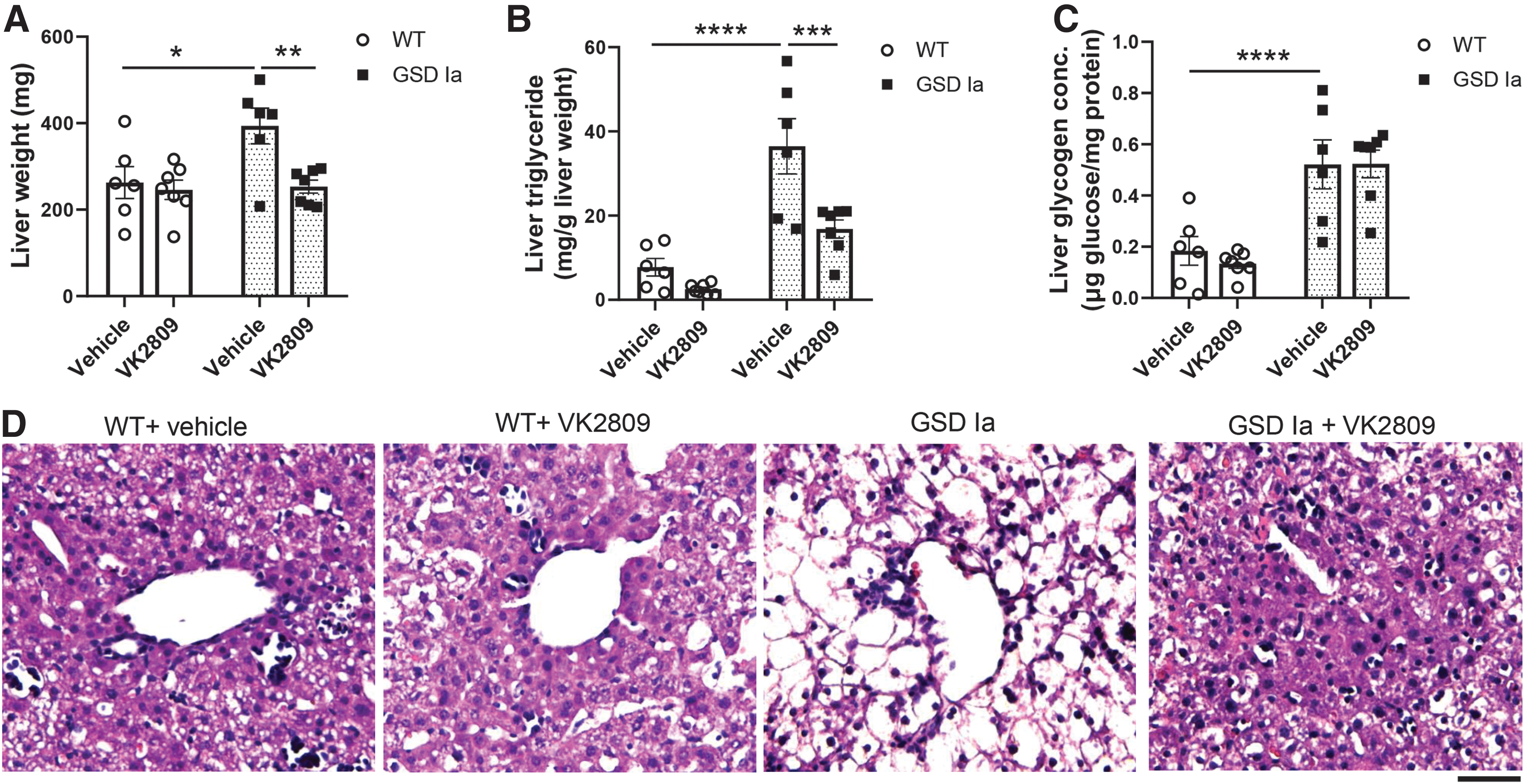
VK2809 treatment decreased hepatic triglyceride and liver mass in GSD Ia mice.
We next assessed the effect of VK2809 on hepatosteatosis by measuring hepatic triglyceride content and found that VK2809 treatment significantly decreased the hepatic triglyceride concentration in the livers of GSD Ia mice (Fig. 1B). However, VK2809 had no significant effect on glycogen concentration in the livers of GSD Ia or WT mice (Fig. 1C). We observed decreased hepatic lipid content and improved hepatic cell morphology by hematoxylin–eosin staining in these GSD Ia mice with VK2809 (Fig. 1D). However, VK2809 treatment did not affect serum glucose and triglyceride levels, or body mass in GSD Ia mice (Supplementary Fig. S1B–D).
VK2809 treatment restores hepatic autophagy in GSD Ia
Autophagy is a critical process that increases β-oxidation of fatty acids by TH in the liver (27), and it is impaired in the livers of GSD Ia mice (8). We found that VK2809 increased the hepatic expression of autophagosome marker, microtubule-associated protein light chain 3-II (LC3B-II), and decreased p62 proteins to levels similar to those observed in WT mice (Fig. 2A–C). P62 is an ubiquitin-binding scaffold protein that is degraded inside the autolysosome, and its level is inversely correlated with autophagic flux (35). Thus, VK2809 treatment restored hepatic autophagosome formation and autophagic flux in GSD Ia mice.
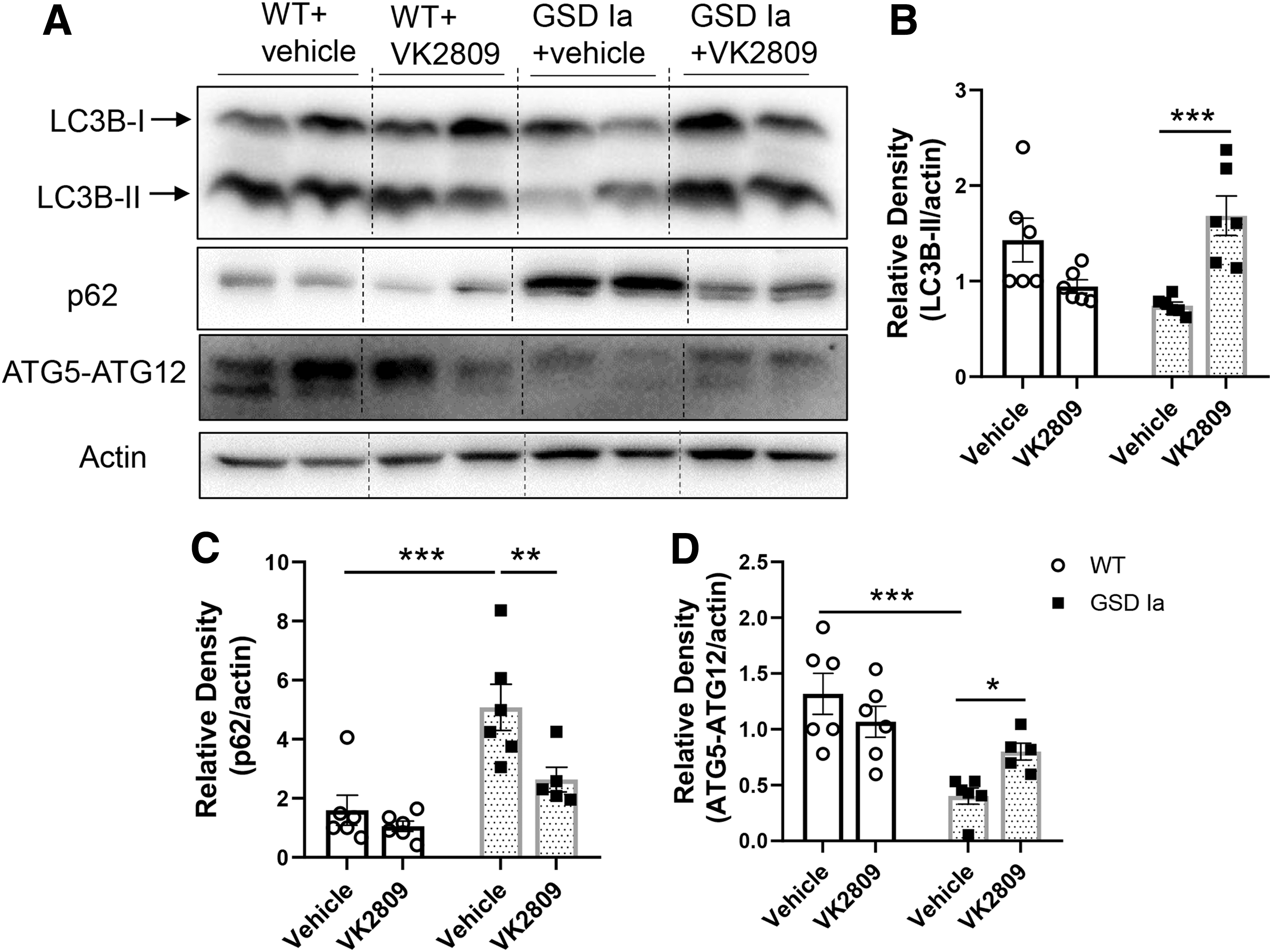
VK2809 increased hepatic autophagy in GSD Ia mice.
We next assessed the impact of VK2809 on autophagy-related (ATG) and lysosomal proteins that comprise the autophagy machinery. ATG5-ATG12 conjugation is critical for the formation of LC3B-II and is downregulated in GSD Ia (8,17). ATG5-ATG12 conjugation was increased by VK2809 in the livers of GSD Ia mice (Fig. 2A, D). Furthermore, VK2809 treatment increased several lysosomal proteins, including lysosomal proteases, cathepsin B and D, lysosomal-associated membrane protein 2 (LAMP2), and ATPase H+ transporting V0 Subunit A2 (ATP6V0A2), a subunit of vacuolar ATPase (v-ATPase) that is essential for lysosomal acidification. Altogether, these results suggest that VK2809 increased lysosomal degradation capacity in GSD Ia mice (Fig. 3).
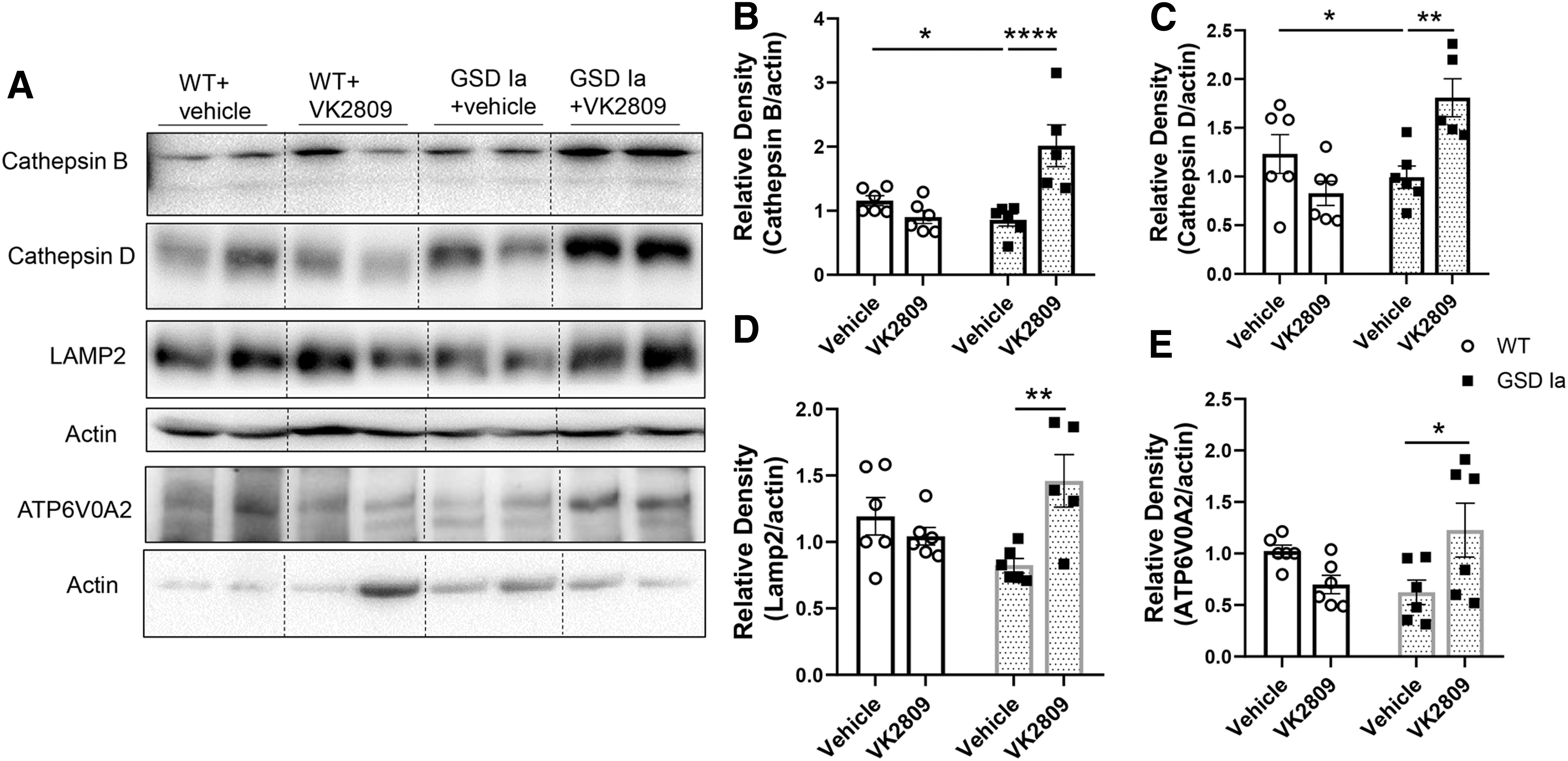
VK2809 treatment increased lysosomal proteins in the livers of GSD Ia mice.
AMP-activated protein kinase (AMPK) and the mammalian target of rapamycin (mTOR) signaling regulate autophagy and are perturbed in GSD Ia (8). Therefore, we assessed the effect of VK2809 treatment on AMPK and mTOR signaling and found that VK2809 decreased phosphorylation of mTOR and its downstream target, eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4EBP1) in GSD Ia mice (Fig. 4A–C), indicating that VK2809 decreased mTOR signaling in the GSD Ia liver. VK2809 treatment also increased the phosphorylations of AMPK and the adipose triglyceride lipase (ATGL) at Ser406 (Fig. 4D–F), a site that is phosphorylated by AMPK (36). These findings indicate that VK2809 increased AMPK signaling as well as decreased mTOR signaling in the livers of GSD Ia mice, which likely led to the restoration of autophagy in the livers of GSD Ia mice.
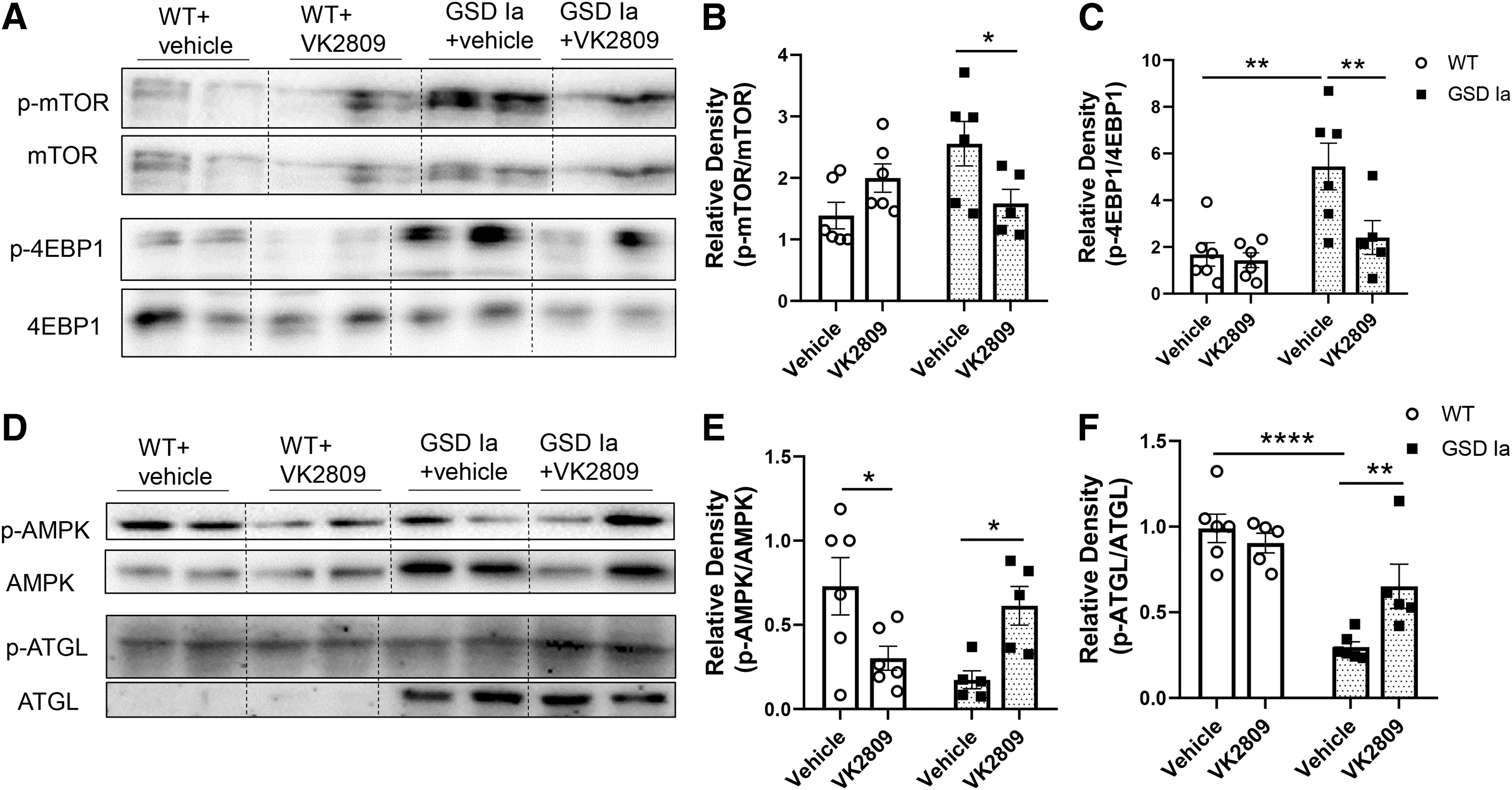
VK2809 treatment decreased mTOR and increased AMPK signaling in the livers of GSD Ia mice. Immunoblots
VK2809 treatment restores mitochondrial biogenesis and β-oxidation in GSD Ia
Dysfunctional mitochondria are key contributors to the pathogenesis of GSD Ia. We previously demonstrated that there is decreased mitochondrial biogenesis and content, impaired oxidative phosphorylation, and increased levels of pyruvate and lactate, as well as altered TCA cycle intermediates in the livers of GSD Ia mice (9). To investigate the effect of VK2809 on mitochondrial biogenesis, we first examined the protein levels of estrogen-related receptor alpha (ERRα) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), key factors that are involved in the regulation of mitochondrial biogenesis (30), and found that VK2809 treatment increased both ERRα and PGC1α protein levels (Fig. 5A–C). VK2809 treatment also increased the levels of four inner and outer membrane mitochondrial proteins: voltage-dependent anion channel 1 (VDAC1), translocase of outer mitochondrial membrane 20 (Tom20), cytochrome c oxidase subunit 4I1 (COXIV), and ATP synthase F1 subunit alpha (ATP5A) (Fig. 5D–H).

VK2809 treatment increased hepatic mitochondrial biogenesis in GSD Ia mice. Immunoblots
TH has been shown to stimulate fatty acid β-oxidation in the liver (15,27,37). In GSD Ia mice, VK2809 treatment increased the protein level of carnitine palmitoyltransferase 1a (CPT1α), a mitochondrial enzyme critical for fatty acid oxidation (Fig. 6A, B). VK2809 also significantly increased the mRNA for fibroblast growth factor 21 (FGF21), a mediator for hepatic lipid catabolism (38) (Fig. 6C). Interestingly, VK2809 treatment did not decrease fatty acid synthase (FAS) level in the livers of GSD Ia mice (Supplementary Fig. S4), suggesting that it may not alter lipogenesis.
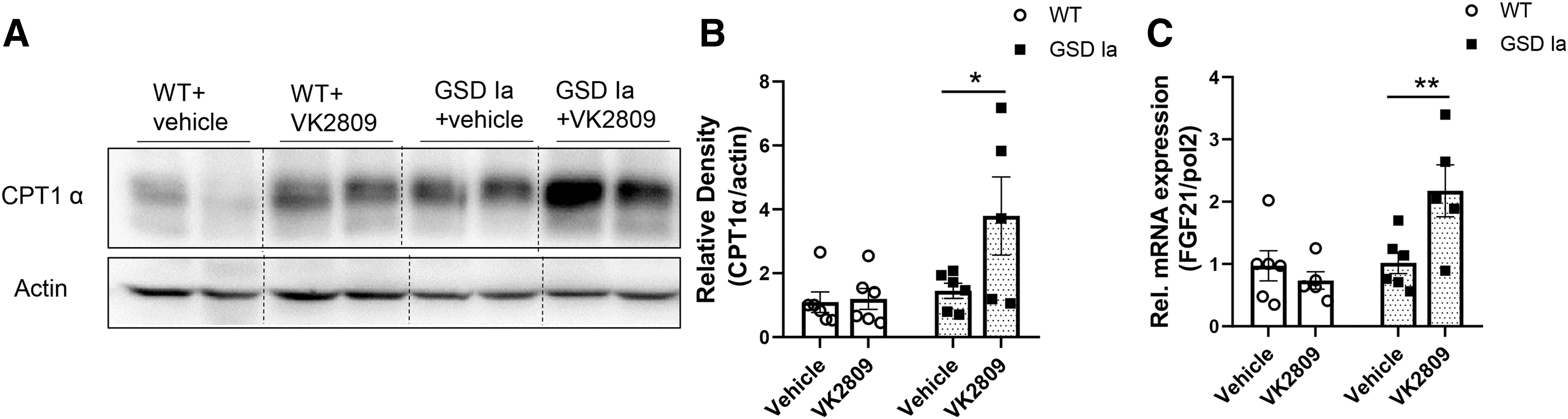
VK2809 treatment increased hepatic CPT1α and FGF21 in GSD Ia mice.
Discussion
In the current study, we treated GSD Ia mice with a liver- and THRβ-specific thyromimetic prodrug, VK2809, and demonstrate that it decreased hepatosteatosis in the livers of GSD Ia mice by increasing fatty acid β-oxidation through the induction of autophagy, mitochondrial biogenesis, and CPT1α expression (Fig. 7). We previously showed that decreased autophagy and mitochondrial dysfunction played prominent roles in the decreased β-oxidation of fatty acids and oxidative phosphorylation seen in GSD Ia hepatic cells (8,9,18). There also is accumulating evidence suggesting that perturbed lipid metabolism significantly contributes to the long-term hepatic complications in GSD Ia, thus reducing hepatosteatosis may be a novel therapeutic strategy for GSD Ia (8,18,39,40).

Prodrug VK2809 is processed in the liver and effect through the activation of THRβ. VK2809 (formerly known as MB07811) is a prodrug, which undergoes first-pass hepatic extraction and requires cytochrome P450 cleavage to generate the negatively charged THRβ agonist. This THRβ agonist decreases hepatosteatosis in the livers from GSD Ia mice by increasing fatty acid β-oxidation through the induction of autophagy, mitochondrial biogenesis, and enzymes for lipid oxidation. THRβ, thyroid hormone receptor β.
VK2809 treatment restored the impaired hepatic autophagy flux in GSD Ia mice as evidenced by increased LC3B-II and decreased p62 levels (Fig. 2). Stimulation of autophagy of lipid stored in fat droplets (lipophagy) reduces hepatosteatosis. The hydrolysis of triglycerides into free fatty acids that occurs after autophagosome/lysosome fusion generates the fuel that is catabolized by mitochondrial β-oxidation (15,27,37). TH increases autophagy by regulating AMPK or/and mTOR signaling, increasing ATG protein expression, and stimulating lysosomal activity (28,29,31,41). The formation of autophagosomes is impaired in GSD Ia, most likely related to altered cellular signaling and decreased levels of ATG proteins (8,17). In GSD Ia mice, VK2809 increased AMPK phosphorylation, reduced mTOR phosphorylation (Fig. 4), increased ATG5-ATG12 conjugation (Fig. 2), and increased the expression of lysosomal proteins, such as LAMP2, cathepsin B, cathepsin D, and ATP6V0A2 (Fig. 3). Interestingly, we did not observe any decrease in glycogen content in this short-term model, despite induction of autophagy. VK2809 may have stimulated lipophagy but not glycophagy at the dose used. However, it also is possible that VK2809 induced glycogen synthase activity in addition to promoting autophagy-dependent degradation. Malbon and Campbell demonstrated that in vivo administration of triiodothyronine (T3) or T4 rapidly increased the enzymatic activity of glycogen synthase (42). Another possibility is that the elevated intrahepatic G6P concentrations in GSD Ia mice activate glycogen synthase and block the phosphorylation of glycogen synthase by protein kinase A (43) to mask any effect of VK2809 on glycogen stores by glycophagy.
We previously found that rapamycin (7) and a pan-peroxisome proliferator-activated receptor (PPAR) agonist, bezafibrate (18), markedly reduce hepatosteatosis in GSD Ia mice through simulation of autophagy and β-oxidation of fatty acids. Thus, the simultaneous induction of autophagy and β-oxidation enzymes such as CPT1α may be indispensable for the lipid-lowering effect by VK2809 in GSD Ia. The ability to restore autophagy, to enhance mitochondrial β-oxidation, and to stimulate mitochondrial biogenesis (see below) makes PPAR or THRβ agonists attractive agents to treat hepatosteatosis in GSD Ia or NAFLD. An earlier study demonstrated sirtuin 1 (SIRT1)-dependent dysregulation of autophagy in GSD Ia; however, while genetic restoration of SIRT1 level increased hepatic autophagy, it did not reduce hepatosteatosis (17). These findings suggest that, in addition to its effects on autophagy, the beneficial effects of VK2809 on reducing hepatosteatosis may be due to cellular effects, such as increasing CPT1α expression and mitochondrial biogenesis (Figs. 5 and 6).
Recent studies have shown that there is impaired fatty acid β-oxidation and respiration, altered metabolite levels (e.g., acylcarnitines), and decreased autophagy and biogenesis of mitochondria in GSD Ia (9). Mitochondrial dysfunction and associated changes in hepatic lipid metabolism play important roles in type 2 diabetes or NAFLD; thus, it is likely they also contribute to the pathogenesis of GSD Ia. Mitochondrial biogenesis can be induced by several nuclear receptors, including THR (28 –30). We recently showed that TH increased mitochondrial biogenesis through upregulation of the key cofactors PGC1α and ERRα (30). In the GSD Ia livers, we found that VK2809 treatment increased the protein levels of PGC1α and ERRα and increased the hepatic levels of mitochondrial proteins, such as VDAC1, Tom20, ATP5A, and COXIV (Fig. 5). Taken together, these results show that VK2809, similar to TH, reduces hepatic triglyceride content by stimulating fatty acid oxidation and increasing mitochondria biogenesis.
We previously reported that TH treatment increases hepatic autophagy and mitochondrial biogenesis (27,28,30). However, in the current study, we noticed that VK2809 treatment increases autophagy, and mitochondrial and lysosomal proteins in GSD Ia mice but not in WT mice. It is possible that the age of the studied mice plays a role in these differences. Our previous studies used 2- to 3-month-old adult mice, while the current study used 5-day-old mice. It has been reported that there is a rapid increase in serum T4 level postnatally that occurs from day 0 to day 7 (34). Thus, it is possible that the optimal dose of VK2809 to increase hepatic autophagy and mitochondrial biogenesis may be different for adult than newborn mice. VK2809 is a prodrug that requires cytochrome P450 cleavage to generate the negatively charged THRβ agonist, VK2809A (formerly known as MB07344) (25); however, little is known about the cytochrome P450 activity in newborn mice or GSD Ia. Differential expression in p450 expression of newborn or GSD Ia mice may explain the observed discrepancy. Studies in patients with NAFLD suggest that there is altered expression and activity of cytochrome P450 (44). Thus, it is possible that GSD Ia, age, or NAFLD may have altered hepatic cytochrome P450 activity that leads to a different intrahepatic VK2809A concentration, which then changes the biological effects of VK2809 observed between the GSD Ia and WT mice in our study.
Recently, low-dose levothyroxine supplementation with careful titration decreased hepatosteatosis in diabetic male patients with little or no significant adverse effects (20). However, a major concern for the clinical use of TH and THRβ-selective mimetics for liver disease is their potential adverse effects on other organs, such as the heart and bone. Several strategies have been employed to optimize the effects on the liver while minimizing the side effects on other tissues. THRβ is the predominant TH receptor isoform in the liver, while THRα is the predominant isoform in the heart and bone. In this context, a THRβ-specific thyromimetic MGL-3196, which originally was developed for the treatment of hypercholesterolemia (45), was effective in reducing hepatosteatosis in a phase 2 trial for biopsy-proven nonalcoholic steatohepatitis in preliminary studies (
Footnotes
Acknowledgments
The authors would like to acknowledge inspiration and support from Dr. Emory and Mrs. Mary Chapman and their son Christopher, and from Dr. and Mrs. John Kelly. We deeply appreciate the dedication shown by the staff of the Duke Department of Laboratory Animal Resources, as well as undergraduate students at the Duke University. We thank Dr. Janice Chou at the National Institute of Child Health and Human Development for providing G6pc −/− mice.
Authors' Contributions
J.Z., L.R.W., D.D.K., and P.M.Y. conceived the experiments. J.Z., L.R.W., A.L., and X.-H.L. performed the experiments and analyzed the data. J.Z., P.M.Y., and D.D.K. wrote the article. All authors provided critical feedback and helped shape the research, analysis, and article.
Author Disclosure Statement
D.D.K. served on a data and safety monitoring board for Baxter International. He was supported by a grant from Roivant Rare Diseases. He received an honorarium and grant support in the past from Genzyme Sanofi. He holds equity in Actus Therapeutics. D.D.K. and P.M.Y. have developed the technology that is being used in the study. If the technology is commercially successful in the future, the developers and the Duke University may benefit financially.
Funding Information
This work was supported by Singapore NMRC grant NMRC/CIRG/1457/2016 and NRMC/CSA/0054/2013 to P.M.Y., and grant R01DK105434-01A1 from the National Institute of Diabetes and Digestive and Kidney Diseases, as well as by support from the Alice and Y. T. Chen Center for Genetics and Genomics to D.D.K.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4