
Abstract
Background:
Genetic alterations activating the mitogen-activated protein kinase (MAPK) signaling pathway, most commonly BRAF and RAS mutations, are common in papillary thyroid carcinoma (PTC). Somatic mutations of the MEK gene, also known as mitogen-activated protein kinase kinase 1 (MAP2K1), coding for a signaling protein downstream of BRAF, have been found in several cancer types. The goal of this study was to investigate if functional MEK1 mutations occur in thyroid cancer (TC).
Methods:
We analyzed MEK1 mutation status in a series of 101 PTCs lacking other known driver mutations using Sanger sequencing and targeted next-generation sequencing. In addition, 64 follicular and Hürthle cell carcinomas and 32 follicular adenomas were studied. The occurrence of MEK1 mutations was evaluated using another series of thyroid tumors studied by targeted next-generation sequencing. Western blot and RNA-seq analyses were performed on selected tumors.
Results:
We detected MEK1 mutations in 2/101 (2%) PTCs that otherwise had no known genetic alterations, in 0/64 follicular and Hürthle cell carcinomas, and in 0/32 follicular adenomas. Two positive tumors carried the same in-frame deletion p.L98_I103del; K104I (c.292_309del18; c.311A>T) located in exon 3 of the gene. One additional MEK1 mutation was identified following routine molecular tumor profiling. The tumor had an in-frame deletion p.I99_K104del (c.294_311del18) also located in exon 3. Western blot analysis of one of the tumors showed activation of the MAPK pathway. Using RNA-seq analysis to evaluate changes in gene expression, these tumors were RAS-like and showed a high thyroid differentiation score. Phenotypically, the MEK1-positive PTCs were all encapsulated and had a predominantly or exclusively follicular architecture, being diagnosed as a classic papillary type with a significant follicular pattern ( × 2) or follicular variant PTC ( × 1). Follow-up was available for 2 patients, with no evidence of disease found 2 and 10 years postsurgery.
Conclusions:
In this study, we report the occurrence of functional MEK1 mutations in PTC. All mutations are in-frame deletions in exon 3 of MEK1, representing another mechanism of activation of the MAPK pathway in papillary carcinomas with a predominantly follicular growth pattern.
Introduction
Thyroid cancer (TC) is the most common endocrine malignancy. It constitutes ∼3% of all newly diagnosed cancer cases in the United States (1) and 2% of all newly diagnosed cancer cases worldwide (2). Papillary thyroid carcinoma (PTC) is the most common type of thyroid malignancy, which accounts for close to 80% of TCs in adults, followed by follicular carcinoma and Hürthle cell carcinoma (3 –5).
The molecular pathogenesis of papillary carcinomas involves activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase/AKT signaling pathways. The most frequent alterations leading to the activation of the MAPK pathway are point mutations of the BRAF and RAS genes and rearrangements of RET, BRAF, NTRK3, NTRK1, ALK, THADA, and other genes (6 –10). The Cancer Genome Atlas (TCGA) study, which provided a broad genomic analysis of >400 cases of PTC (7), detected nonoverlapping point mutations in ∼75% and chromosomal rearrangements in ∼15% of these tumors (7). Even if it is accepted that the recurrent DNA copy number alterations that were identified by TCGA in ∼7% of PTC are driver events, a small but distinct fraction of PTCs lacked a driver molecular alteration. This suggests that additional genetic mechanisms, likely leading to activation of the MAPK pathway, may exist in PTC tumorigenesis.
The primary downstream effector of BRAF is MEK1. The MEK1 gene, also known as mitogen-activated protein kinase kinase 1 (MAP2K1), is located on chromosome 15 and encodes MEK1 protein kinase. Once activated, MEK1 catalyzes the phosphorylation of threonine and tyrosine residues of ERK (11). Deletions and mutations in the activation segment of MEK1 have been shown to constitutively activate the protein (12,13). Mutations of MEK1 have been identified in various types of human cancer, including lung, melanoma, colon, Langerhans cell histiocytosis (LCH), and hairy cell leukemia (14 –19). The majority of previously reported MEK1 mutations occur in the autoregulatory domain encoded by exon 2 and the catalytic core encoded by exon 3. Almost all of these mutations cause constitutive activation of the MEK1 kinase (15,18,20). The most prevalent mutations detected were missense, except for LCH in which many mutations were in-frame deletions (17,19). MEK1 mutations were found almost always to be mutually exclusive with other driver mutations (14,17,19), except for melanoma where they often occurred together with BRAF or NRAS mutations (21,22).
The aim of this study was to determine the prevalence of MEK1 mutations in PTC negative for other common driver mutations and investigate functional consequences and histopathologic features of thyroid tumors harboring mutations in the MEK1 gene.
Materials and Methods
Patient samples
With the approval of the University of Pittsburgh Institutional Review Board, a series of 110 consecutive cases of PTC from the Department of Pathology, University of Pittsburgh, previously found to have no common driver mutations, including BRAF, NRAS, HRAS, KRAS, RET/PTC, ETV6-NTRK3, EML4-ALK, STRN-ALK, and THADA-IGF2BP3, were analyzed. We also studied 24 follicular thyroid carcinomas, 40 Hürthle cell carcinomas, and 32 follicular thyroid adenomas. In addition, one MEK1 mutation-positive case was identified at the Department of Pathology, Brigham and Women's Hospital, during routine molecular testing via the OncoPanel assay that analyzes exonic DNA sequences of 447 cancer genes and 191 regions across 60 genes for rearrangements as described previously (23,24). This case was recently reported (25).
MEK1 mutational analysis
Genetic analysis was performed on formalin-fixed and paraffin-embedded or snap-frozen tissue samples. DNA was purified using the QIAamp DNA Mini Kit (Qiagen GmbH, Hilden, Germany). MEK1 mutational analysis of exons 2 and 3 was performed using Sanger sequencing. Briefly, DNA (∼80 ng) was amplified using 0.2 μM polymerase chain reaction (PCR) primers flanking exon 2 (forward: 5′-TGACTTGTGCTCCCCACTTT-3′ and reverse: 5′-AGGCAAACTCACCTTTCTGG-3′) and exon 3 (forward: 5′-CATCCCTTCCTCCCTCTTTC-3′ and reverse: 5′-CTCTTAAGGCCATTGCTCCA-3′) in a final volume of 30 μL containing 1 × PCR buffer, 0.1 mM dNTP, 1.5 mM MgCl2, and 2.5 U of AmpliTaq Gold DNA Polymerase (Thermo Fisher Scientific). PCR started at 95°C for 15 minutes, followed by 35 cycles of 94°C for 30 seconds, 57°C for 30 seconds, and 72°C for 30 seconds, and finished by 5 minutes of 72°C.
RNA-sequencing and data analysis
RNA from snap-frozen tissue samples was isolated as previously reported (26). Cluster generation and paired-end sequencing were performed on Illumina HiSeq2500 using the HiSeq Paired-End (PE) Rapid Cluster kit v2 and HiSeq Rapid SBS kit v2 (Illumina). The filtered high-quality reads from the HiSeq sequencing system were aligned to the human genome (hg19-GRCh37) using TopHat aligner (27) and the number of reads mapped to each gene was calculated using RSeM (28) and featureCounts (29) tools. Differential expression analyses were performed by edgeR package (30) using read counts from RSeM. In addition to overall gene expression analysis, the thyroid differentiation score (TDS) and BRAFV600E-RAS score (BRS) were calculated as previously described (7). Specifically, TDS was calculated as the mean log2 normalized fold change of the expression level of 16 thyroid metabolism- and function-related genes compared with the mean expression level of these genes in 391 PTC cases obtained from the TCGA study (7). BRS was calculated using the expression of 71 genes described in the TCGA study.
Western blot analysis
Total protein was isolated from snap-frozen tissue homogenized in RIPA buffer containing protease inhibitors (Roche) and phosphatase inhibitors (Sigma) using OMNI-GLH with hard tissue tips (OMNI International). Protein concentration was determined using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific) according to the manufacturer's protocol, and 40 μg of total protein was fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Lonza) and transferred to the nitrocellulose membrane (Bio-Rad). The following primary and secondary antibodies were used: MEK1/2 antibody (1:1000, cat. no. 9122; Cell Signaling Technology), phospho-MEK1/2 (Ser217/221) (41G9) (1:1000, cat. no. 9154; Cell Signaling Technology), p42/44 MAPK (1:1000, cat. no. 9102; Cell Signaling Technology), phospho-p42/44 MAPK (pT202/pY204) (1:1000, cat. no. 9101; Cell Signaling Technology), and anti-rabbit (1:10,000, cat. no. W401B; Promega).
Pathology review
Pathology reports and glass slides of MEK1 mutation-positive tumors were retrieved and reviewed. Tumors were classified according to the 2017 WHO classification of endocrine tumors (31).
Results
MEK1 mutation analysis
A total of 101 consecutive cases of PTCs lacking all known major driver mutations were studied by Sanger sequencing for mutations in the autoregulatory and catalytic domains encoded by exons 2 and 3 of the MEK1 gene, respectively. Among these samples, two tumors were found positive for mutations. Both MEK1-positive PTCs carried the same in-frame deletion: p.L98_I103del; K104I (c.292_309del18; c.311A>T) (Fig. 1). The mutation occurred in exon 3 within the catalytic domain and resulted in a deletion of 6 codons (18 bp) coding for a leucine (L), isoleucine (I), histidine (H), leucine (L), glutamic acid (E), and isoleucine (I) from positions 98 to 103. In addition, it led to a substitution of a lysine (K) to isoleucine (I) at position 104.
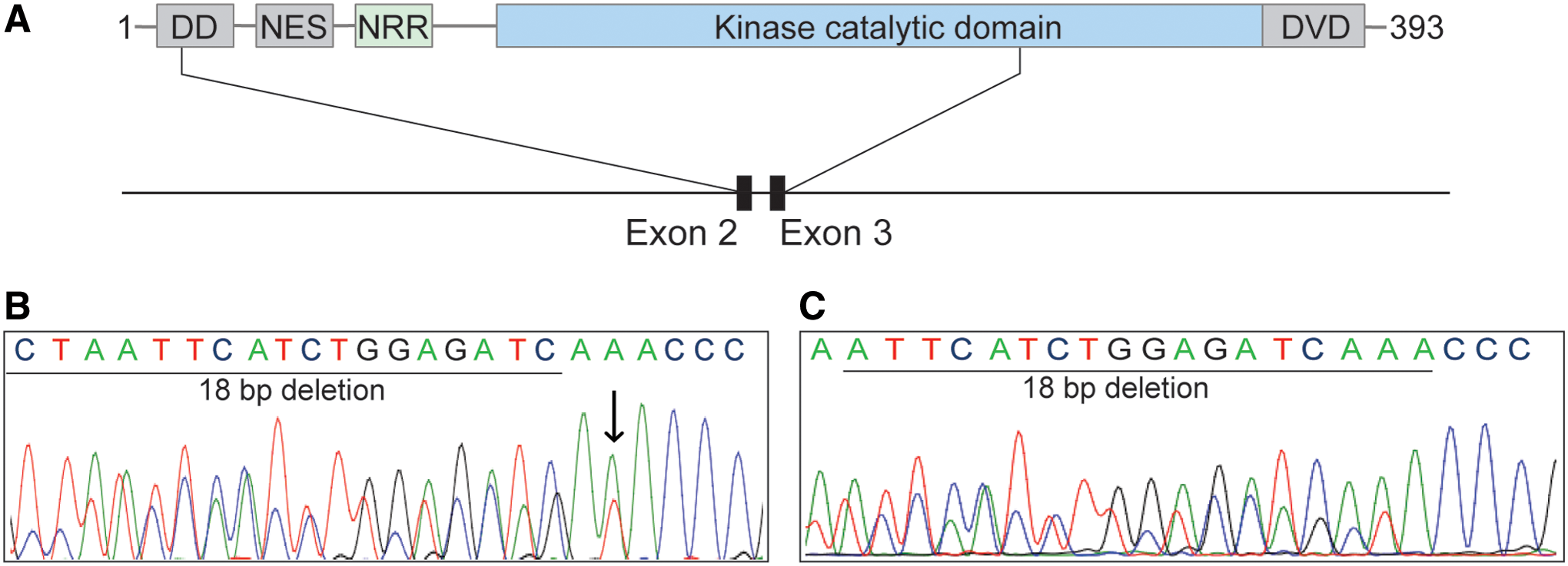
MEK1 mutations in TC. (
Next, we analyzed a series of 40 Hürthle cell carcinomas, 24 follicular carcinomas, and 32 follicular adenomas for MEK1 mutations. None of the tumors was positive for these mutations (Table 1).
Prevalence of MEK1 Mutations in Thyroid Tumors
Selected based on the absence of commonly known driver mutations.
PTC, papillary thyroid carcinoma.
An additional case of papillary carcinoma harboring a mutation of the MEK1 gene was identified by routine targeted next-generation sequencing at the Brigham and Women's Hospital. The mutation p.I99_K104del (c.294_311del18) was an in-frame deletion of 18 bp involving exon 3 of the gene. It was confirmed by Sanger sequencing (Fig. 1). The deletion caused the loss of 6 amino acids: isoleucine (I), histidine (H), leucine (L), glutamic acid (E), isoleucine (I), and lysine (K) from positions 99 to 104 of MEK1. This tumor, evaluated by a next-generation sequencing assay that surveys exonic DNA sequences of 447 cancer genes and 191 regions across 60 genes for rearrangements, did not reveal any other somatic mutations or structural variants.
Western blot analysis
Similar mutations affecting the catalytic core of MEK1 have previously shown to result in constitutive activation of the MAPK pathway (15 –17,19,32). To take advantage of the availability of snap-frozen tissue from one of the tumors harboring the L98_I103del; K104I mutation of MEK1, we used Western blot analysis for the phosphorylation status of MEK and ERK. ERK is the downstream target of MEK in the MAPK signaling cascade. We observed the increase in phosphorylation of MEK and ERK proteins in the tumor compared with normal thyroid tissue, similar to that seen in PTC harboring BRAFV600E or NRASQ61R mutations, indicating that the MAPK signaling is activated in the MEK1-mutated PTC (Fig. 2).

Functional consequences of MEK1 mutation. Western blot analysis of protein lysates from tumor with MEK1 L98_I103del; K104I, BRAFV600E , or NRASQ61R mutations and normal thyroid using antibodies specific for the total protein levels (t-) and phosphorylated forms (p-) of the indicated mitogen-activated protein kinase pathway proteins.
BRAF-RAS score and TDS
To further understand the consequences of the new driver mutation on gene expression profiles of these PTCs, we performed RNA-Seq analysis of the two tumors positive for p.L98_I103del; K104I MEK1 mutation. First, we established a 71-gene signature determining the BRAFV600E-RAS score (BRS), which determines that the expression profile of a given tumor is closed to tumors carrying BRAFV600E mutation or RAS mutation (7). We found that both MEK1-mutated PTCs were RAS-like (BRS score 0.342 and 0.456) (Fig. 3). We further explored the expression signature of 16 genes used in the TCGA study to establish the TDS (7). The analysis revealed a positive TDS (0.256 and 0.553) in both PTCs carrying MEK1 mutation, similar to the RAS-positive samples (Fig. 3). These results indicate that MEK1-mutated PTCs preserve the expression of genes associated with thyroid differentiation and overall have the expression signature more similar to that seen in tumors driven by RAS mutations.
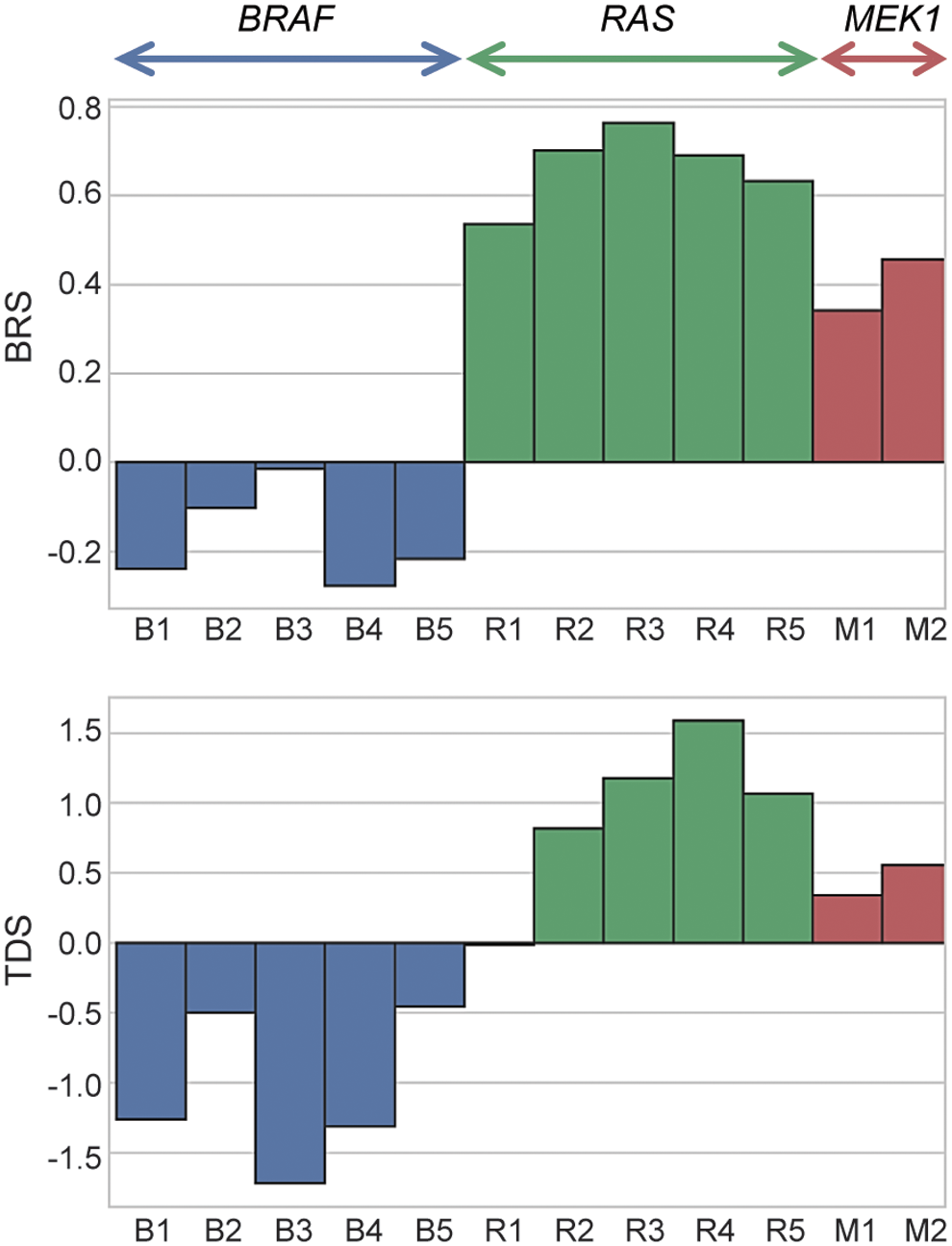
BRS and TDS. Samples were colored by driver gene mutation type (BRAF, RAS, MEK1). Top: The BRS based on the expression of signature of 71 genes. Tumors with negative BRS (−1 to 0) are defined BRAFV600E-like, while tumors with positive BRS (0 to1) are defined as RAS-like. Bottom: TDS based on the expression signature of 16 genes. Negative scores refer to low differentiated tumors and positive scores to well-differentiated tumors. BRS, BRAFV600E-RAS score; TDS, thyroid differentiation score. Color images are available online.
Histopathology and clinical follow-up
The age and sex of patients and size of tumors positive for harboring MEK1 mutations are shown in Table 2. Histopathologic analysis of the surgically excised tumors revealed that all tumors were encapsulated, had a predominantly or exclusively follicular architecture, and showed well-developed nuclear features of papillary carcinoma. Specifically, tumor 1 had a complete thin to moderately thick capsule and showed an almost exclusively follicular architecture, with only several small areas of papillary growth, yielding a diagnosis of classical type PTC (Fig. 4A, B). Tumor 2 had a complete moderately thick capsule and consisted of a mixture of round to elongated follicles and papillary structures (Fig. 4C, D). Tumor 3 had a complete mostly thin capsule and exclusively follicular architecture, with intermingling groups of small and larger size follicles (Fig. 4E, F). In addition to other nuclear features of PTC, this tumor had nuclear pseudoinclusions, which are rarely found in the encapsulated follicular variant of PTC. Capsular invasion was seen in two tumors and no lymphovascular invasion was found (Table 2). All tumors were intrathyroidal. All three patients underwent total thyroidectomy and were treated with radioactive iodine. Follow-up information was available for two of these patients, with no adverse events identified 2 and 10 years after the initial surgery.

Histopathologic features of MEK1 mutation-positive papillary thyroid carcinoma. (
Summary of Thyroid Carcinomas Harboring MEK1 Mutations
cPTC, classical papillary thyroid carcinoma; F, female; FVPTC, follicular variant of papillary thyroid carcinoma; M, male; NED, no evidence of disease; NI, no information.
Discussion
In this study, we demonstrate that MEK1 mutations represent another mechanism of MAPK pathway activation in PTCs.
MEK1 mutations have been reported in several types of tumors (14 –16,33 –35). The majority of previously reported MEK1 missense mutations or deletions targeted exons 2 and 3, which encode the negative regulatory domain and the catalytic core, respectively (20,36). One study investigated the occurrence of MEK1 mutations in TCs including papillary, follicular, and anaplastic carcinomas (34). No MEK1 mutations were identified in that study, possibly because the analysis involved only exon 2 of the gene.
All mutations identified in this study are in-frame deletions located in the kinase domain coded by exon 3. The overall MEK1 mutation rate reported in PTCs by the cBio Cancer Genomics Portal (TCGA; PanCancer Atlas) (
Similar deletions in this region have been described in two cases of LCH and shown to lead to constitutive activation of the MAPK pathway and increasing MEK1 enzymatic activity (19,37). This is consistent with our findings in one of the MEK1 mutation-positive TCs, which showed strong upregulation of ERK, a downstream effector of MEK in the MAPK signaling pathway.
Recently, Yuan et al. (38) characterized the mechanisms of activation caused by several in-frame deletions in the activation loop of MEK1. Specifically, it has been shown that deletions involve the b3-aC loop and enhance MEK1 homodimerization with cross-phosphorylation and constitutive activation of the protein that promotes tumorigenesis.
In this study, all three MEK1-positive tumors were wild type for all other common driver mutations known to occur in TC. This provides additional evidence in favor of MEK1 being a primary driver mutation in these tumors. Of note, MEK1 mutations were found to be mutually exclusive with other driver mutations in most other tumor types such as LCH, hairy cell leukemia, and lung cancer (14,18,19,35). The only well-documented exception is malignant melanoma in which MEK1 mutations frequently coexist with other mutations, including BRAF (16,21,22).
Histopathologically, the MEK1-mutated tumors in this study were papillary carcinomas, which were encapsulated and showed predominantly or exclusively a follicular growth pattern. However, we did not find these mutations among 64 follicular and Hürthle cell carcinomas and 32 follicular adenomas that we analyzed. It is well established that the most frequent genetic alteration identified in PTCs is BRAFV600E . This mutation is commonly found in the classic papillary type of PTC, and is usually associated with more aggressive phenotypic features such as absence of capsule, lymph node metastases, and advanced stage (7,39,40). Alternatively, follicular variants of PTCs frequently harbor RAS mutations and may have a BRAFK601E mutation with lower prevalence, and they are usually associated with tumor encapsulation and frequently an indolent behavior (9,40,41). It is also established that these two main mutually exclusive drivers, BRAFV600E and RAS, show distinct gene expression signatures that can be used to formulate a BRAFV600E-RAS score. This score is based on the differential expression of the 71 genes showing the largest difference in expression levels between the two groups (7). Interestingly, the two MEK1-positive tumors that we evaluated in this study had an RAS-like score. Although this could be considered unexpected since MEK1 is located directly downstream of BRAF in the MAPK signaling cascade, it is important to note that whereas BRAFV600E tumors have a gene expression signature yielding a BRAF-like score, tumors with BRAFK601E are RAS-like (7). Furthermore, the phenotypical tumor features, and specifically encapsulation and predominant follicular architecture, correlate well with the observed RAS-like score.
Despite the relatively large size of these tumors, all of them were encapsulated and showed only limited tumor capsule invasion and no vascular invasion or extrathyroidal extension. Furthermore, these tumors were well differentiated histopathologically and showed a high TDS, which is typically seen in RAS-like tumors (7). On limited follow-up, no adverse events were registered. These findings suggest that overall the MEK1 mutation-positive papillary carcinomas are more likely to be low-risk tumors.
In this study, we also analyzed the negative regulatory domain of MEK1, encoded by exon 2, which is frequently targeted by somatic mutations in other tumors (16 –18,20). No somatic mutations were identified in the 197 thyroid tumor samples that we analyzed. Even if we cannot exclude the presence of mutations in this negative regulatory domain, our results support a previous observation of the absence of exon 2 MEK1 mutations in TC (34).
In summary, this study reports MEK1 mutation as a novel driver event in TC and shows that these tumors are likely to have biological, phenotypical, and clinical characteristics similar to other RAS-like PTCs.
Footnotes
Author Disclosure Statement
M.N.N. is a consultant for Loxo Oncology. For all other authors, no competing financial interests exist.
Funding Information
Supported in part by the National Institutes of Health Grant R01 CA181150.