
Abstract
Background:
Evidence for transgenerational epigenetic inheritance in humans is still controversial, given the requirement to demonstrate persistence of the phenotype across three generations. A previous study showed that exposure of human and mouse embryos to high maternal thyroid hormone (TH) concentrations not only affects the newborns but also subsequently reduces thyrotroph sensitivity to TH during adult life. The current investigation set out to determine if this epigenetic effect is transmitted by humans not exposed in utero to high TH levels to their offspring.
Methods:
The study took advantage of the high frequency of intrauterine exposure to high TH in the Azorean wild-type population born to healthy mothers with high TH levels because of a heterozygous TH receptor beta gene mutation. Wild-type individuals from F2 (second) and F3 (third) generations were studied, whose parents and grandparents, respectively, were not exposed to high maternal TH levels. Twenty-six individuals belonging to 17 nuclear families were tested for their sensitivity to TH using their thyrotropin (TSH) response to thyrotropin-releasing hormone (TRH) after administration of liothyronine (LT3).
Results:
Preservation of reduced sensitivity to TH (RSTH) was found in descendants of males but not of females with likewise RSTH. In F2, offspring of fathers but not of mothers exposed to high TH levels had RSTH (TRH-stimulated TSH of 6.39 ± 0.63 vs 1.58 ± 0.41 mIU/L [p < 0.001], respectively, after treatment with LT3). In F3, whose parents nor themselves were exposed to TH excess during their fetal life, descendants of fathers and not mothers had RSTH (TRH-stimulated TSH of 4.60 ± 0.61 vs 1.37 ± 0.23 mIU/L [p < 0.01], respectively, after pretreatment with LT3).
Conclusions:
Since intrauterine total body and gonadal exposure to elevated TH can potentially affect only the F1 and F2, respectively, the results obtained from F3 confirm a true inheritance of an epigenetic effect, scarcely observed in humans. While the exact mechanism underlying the inheritance of this epigenetic effect remains unknown, it correlates with type 3 deiodinase overexpression demonstrated in pituitary glands of mice born to dams with high TH. This enzyme inactivates TH, and is encoded by an imprinted gene with specific parent of origin expression.
Introduction
The availability of several generations descendent to mothers with resistance to thyroid hormone beta (RTHβ) caused by dominantly inherited mutations in the thyroid hormone receptor beta (THRB) gene, provided a rare opportunity to investigate the effect of exposure to high thyroid hormone (TH) levels during fetal life in individuals not carrying the mutation. Unlike other forms of in utero exposure to TH excess, this TH effect is direct, without consequences secondary to maternal hyperthyroidism, because mothers with RTHβ are shielded from the metabolic effect of hormonal excess (1). Further, due to the dominant transmission of the syndrome, half of the progeny of mothers with RTHβ do not inherit the genetic defect and are thus not protected from the toxic effect of TH excess during their embryonic life. Using this model, it was demonstrated that intrauterine exposure to high maternal levels of TH had both short- and long-term effects on the wild-type (WT; without THRB mutation) progeny of mothers with RTHβ. In contrast, the progeny of fathers with RTHβ did not display these consequences, as their unaffected mothers had normal TH levels. Only WT infants born to mothers with RTHβ had hyperthyroid features such as low birth weight and suppressed thyrotropin (TSH) (2). In the longer term, during adulthood, the stimulation of TSH secretion of these WT individuals, born to mothers with high TH levels, was found to be less suppressible by administration of the biologically active TH liothryonine (LT3), indicating reduced sensitivity to thyroid hormone (RSTH) (3). The finding is consistent with fetal hyperthyroidism having lifelong effects on the feedback regulation of the hypothalamic–pituitary–thyroid axis. Studies in a mouse model of RTHβ showed that the RSTH was associated with increased expression of the pituitary deiodinase type 3 (Dio3) gene, which encodes a membrane selenoenzyme that inactivates TH (4). This observation provided a putative mechanism for the development of RSTH through local reduction of TH-mediated feedback suppression of TSH. As DIO3 is imprinted (5,6), it was wondered if this apparent epigenetic effect is transgenerational—an outcome rarely confirmed in humans (7). The combination of RSTH, being readily demonstrable by reduced TH-induced suppression of the TSH response to thyrotropin-releasing hormone (TRH), coupled with the availability of a large, formerly genotyped Azorean population with many individuals harboring the THRB gene mutation R243Q (2), allowed the inheritance of this epigenetic phenomenon to be followed by studying four generations (F0 through F3).
Methods
The present work took advantage of the relatively frequent occurrence of fetal exposure to high TH levels in the Azorean island of São Miguel. This is the consequence of a missense mutation in the THRB gene causing the amino-acid replacement R243Q, resulting in reduced affinity of the TH receptor beta (TRβ) for TH and thus RTHβ. Its origin has been traced to a couple who lived at the end of the 19th century. F0 represented the third generation and F3 the sixth and seventh generation descendent. In total, 26 individuals belonging to 17 nuclear families were tested for their sensitivity to TH using the magnitude of TSH response to TRH following the administration of LT3. Only individuals of F1, born to mothers with RTHβ, were exposed to high TH levels during embryonic life. Of the eight F1 individuals studied, four were born to two women with RTHβ, and four were the progeny of four men with RTHβ. Four of the F2 individuals were descendants of three women with RSTH, and seven were the progeny of four males with RSTH. All were from F1 parents but not the same F1 individuals studied. In F3, three were the progeny of two women with RSTH, and four were from men with RSTH. All were from F2 but not children of F2 individuals studied. There was no consanguinity among the parents of all individuals studied, and spouses were not related to the original couple with RTHβ.
TRβ R243Q causes increased levels of TH but not thyrotoxicosis (2). Wild-type adults were studied (without THRB gene mutations, first, second, and third generations), descendent of mothers, grandmothers, and great grandmothers with the mutant THβ. Only individuals of generation F1, born to mothers with RTHβ, were exposed to high TH levels during fetal life.
A standard test was used to assess the sensitivity to TH. Each subject received 200 μg TRH intravenously after three days of treatment with TH in the form of liothyronine (LT3) taken orally 25 μg twice daily. Blood was collected at time intervals up to 180 minutes and analyzed for TSH and prolactin. Serum thyroxine (T4), triiodothyronine (T3), reverse triiodothyronine (rT3), sex hormone–binding globulin, ferritin, alkaline phosphatase, cholesterol, and creatinine kinase were also measured before and after treatment. The TRH stimulation test was carried out 12 hours after ingestion of the last dose of LT3. The study was approved by the Institutional Review Boards of Hospital Divino Espírito Santo and The University of Chicago.
Hormones measurements
Serum substances listed above were measured by standard laboratory techniques, as previously detailed (8). Total T4, T3, TSH, and prolactin (PRL) were measured by chemiluminescence immunometric assays using the Elecsys Automated System (Roche, Indianapolis, IN). Total rT3 was measured by radioimmunoassay (Adaltis Italia S.p.A, Bologna, Italy), and thyroglobulin by an in-house radioimmunoassay. Serum fT4 index (fT4I) was calculated as the product of the total serum T4 concentrations and the normalized resin T4 uptake ratio.
Statistical analysis
All results are expressed as the mean ± standard error of the mean. Statistical analysis of multiple groups was by two-way analysis of variance. Student's t-test was used when there were only two groups to compare. p-Values of <0.05 was considered to be significant.
Results
Thyroid tests at baseline were in the reference range in all groups (Table 1), as reported previously for the first generation (F1) by Srichomkwun et al. (3). After LT3 treatment, there was, as expected, a significant increase in serum T3 with a decline in serum T4, fT4I, and TSH that was not statistically different between the group of normo-responsive subjects and those with RSTH. There was no significant age difference between groups of the same generation (Table 1).
Thyroid Function in Progeny of Parents and Grandparents Exposed In Utero to High TH Levels
All results are the mean ± standard error of the mean. Statistically significant values are shown in bold.
Stimulated with thyrotropin-releasing hormone.
TSH, thyrotropin; LT3, liothyronine; PRL, prolactin; T3, triiodothyronine; rT3, reverse T3; T4, thyroxine; fT4I, free T4 index; RSTH, reduced sensitivity to thyroid hormone; NS, not significant (p > 0.05).
Following the administration of LT3, WT individuals born to mothers with RTHβ, and thus exposed in utero to high TH levels, had RSTH as evidenced by a lesser suppression of the increase in serum TSH in response to TRH. The average TSH peak in the F1 born to mothers with RTHβ was 6.8 ± 1.0 compared to 1.6 ± 0.5 mIU/L for offspring of fathers with RTHβ (p < 0.005). There was no overlap in the TSH responses to TRH between the two groups (Fig. 1A). The peak TSH value using this protocol is normally ≤3 mIU/L (1). F2 consisted of descendants of grandmothers with RTHβ. Their parents were exposed to TH excess, but they themselves were not. In this F2, the offspring of fathers but not of mothers exposed to high TH levels had RSTH, as they exhibited a significantly less suppressed peak TSH response to TRH of 6.39 ± 0.63 compared to 1.58 ± 0.41 mIU/L (p < 0.001), respectively, after treatment with LT3 (Fig. 1B and Table 1). F3 was comprised of descendants from great-grandmothers with RTHβ. Neither their parents nor themselves were exposed to TH excess during their fetal life. Similarly to F2, the offspring of fathers but not of mothers had RSTH, as evidenced by lesser reduction of the TRH-stimulated TSH of 4.60 ± 0.61 compared to 1.37 ± 0.23 mIU/L (p < 0.01), respectively, after pretreatment with LT3 (Fig. 1C and Table 1). The slightly lower mean peak TSH response to TRH in F3, offspring of F2 fathers with RSTH, was not significantly different (p = 0.834) from the corresponding F2 group, offspring of fathers exposed to high TH (F1). There was, however, a significant difference (p = 0.045) in age between the two groups (Table 1).
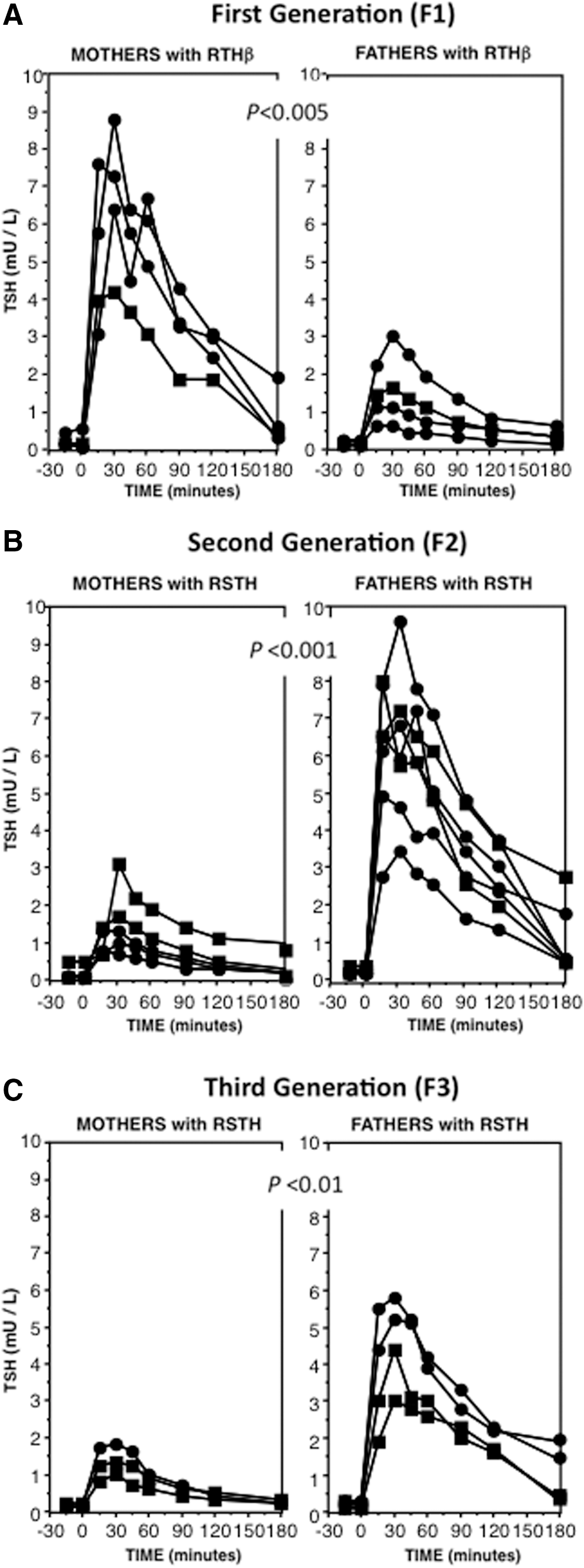
Sensitivity to thyroid hormone (TH) in subjects exposed during fetal life to high levels of TH (F1) and their progeny (F2 and F3)
The PRL response to TRH was not different between the groups (Table 1). No significant differences were observed between the groups in both basal and post LT3 serum concentrations of TSH, T4, T3, rT3, and fT4I, or in the ratios of rT3/T3 and T4/T3 (Table 1). Further, no significant differences between the groups were found for markers of peripheral tissue action of TH, including serum sex hormone–binding globulin, ferritin, alkaline phosphatase, cholesterol, and creatine phosphokinase (data not shown). Of note, the progeny of F1 and F2 who did not manifest RSTH also responded normally to the suppression of the TRH-stimulated TSH following treatment with LT3 (Fig. 2).
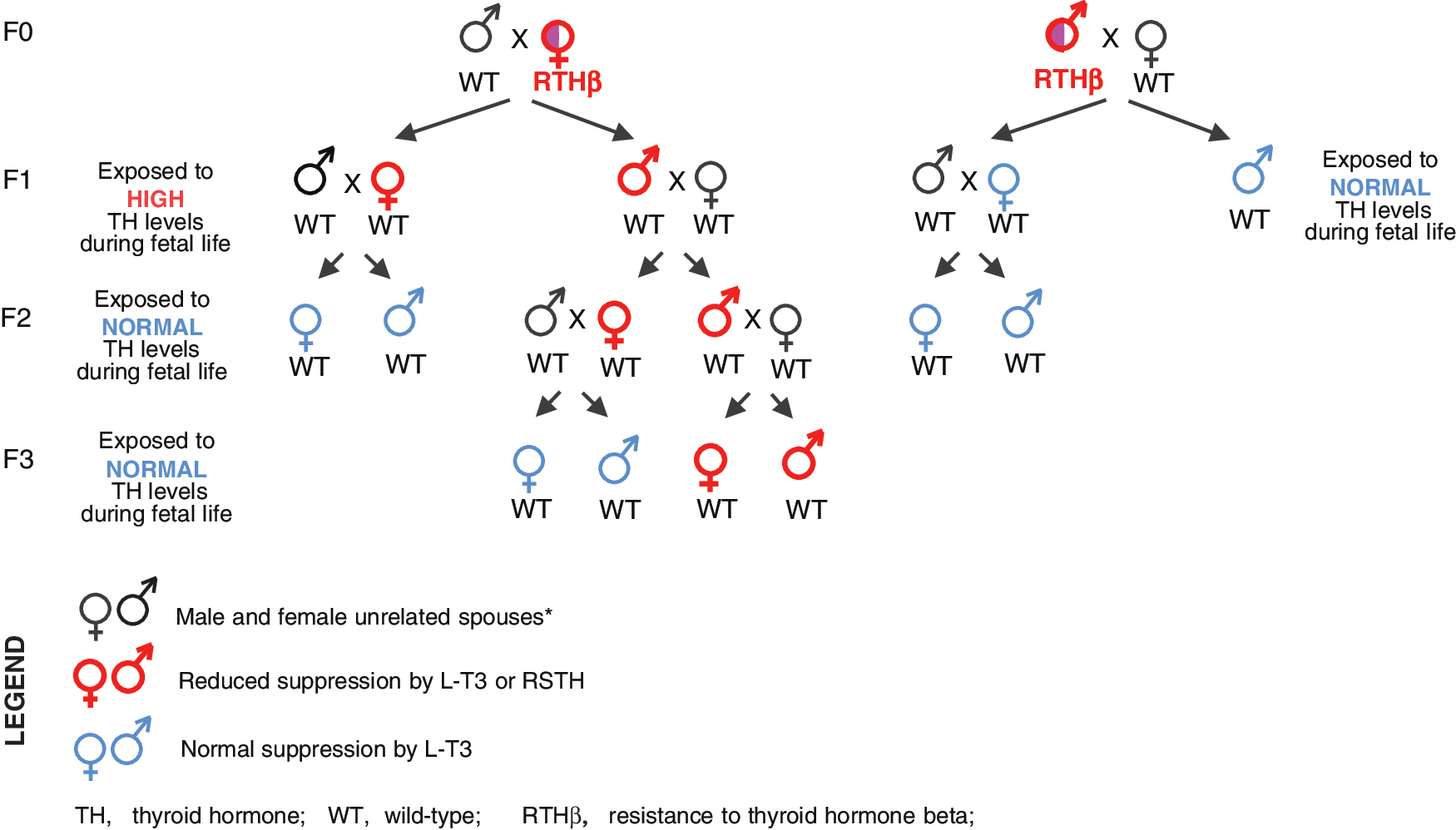
Schematic representation of the genotypes, status of the mates, progeny, and their sensitivity to TH. The transgenerational inheritance of the epigenetic effect producing RSTH along a male line is evident. F1, F2, and F3, generations 1, 2, and 3. *Unrelated to the founders five generations back, except for one spouse each in F1 and F2.
Discussion
The present work demonstrates that fetal exposure to high TH levels causes permanent RSTH that is transmitted by males but not females with RSTH to two subsequent generations of individuals of both sexes not exposed to high TH levels during fetal life (summarized in Fig. 2). The effect appears to be thyrotroph cell-specific, as no alterations were observed in PRL, a hormone secreted by cells that respond TRH with lesser suppression by TH (9). Given the age of individuals of F3 (Table 1), testing for the further transmission awaits maturation to reproductive age. However, since intrauterine total body and gonadal exposure to elevated TH can potentially affect only the F1 and F2, respectively, the results obtained from F3 confirm a true inheritance of an epigenetic effect, scarcely observed in humans (7,10). An earlier study on the transgenerational epigenetic effect of lead exposure inheritance was limited to grandchildren and females exposed to lead and did not extend to great grandchildren representing F3 (11).
The mechanism underlying this transgenerational epigenetic inheritance is not fully understood. It would require incomplete erasure of the parental epigenetic marks in the gametes to allow the transmission of the phenotype across multiple generations. This is known to occur in imprinted genes. Thus, the imprinted DIO3 gene seems to be a fitting candidate to mediate this effect. First, DIO3 inactivates TH and thus regulates the local availability of active TH in a tissue specific manner (12). Second, pituitary Dio3 mRNA was found to be increased in adult mice exposed to high levels of TH during fetal life (3). Third, DIO3 is predominantly expressed from the paternal allele, particularly in the central nervous system (5). Methylation of the imprinting control region of the paternal allele actually enhances DIO3 expression by preventing gene repression through insulator binding (13). Resurgence of epimutations after erasure from one generation to another was shown in three generations of a family with a rare form of young-onset cancer, suggesting that in addition to DNA methylation, other mechanisms such as RNA transcriptional silencing may play a role in the transgenerational inheritance of epigenetic phenomena (14). A very recent report showed that in F2 mice born to males exposed to high TH levels affected genes involved in early brain development through an epigenetic effect on the paternal germline (15). However, true transgenerational inheritance awaits confirmation through the study of F3.
In conclusion, this study demonstrates a transgenerational epigenetic inheritance in humans produced by exposure to high TH in fetal life, in the absence of maternal influences secondary to thyrotoxicosis. The inheritance is along the male line. Based on preliminary animal work, it is postulated that this effect may be mediated by a cell-specific increase in DIO3 expression, an enzyme that inactivates TH, resulting in reduction of its action. It remains unclear whether this resistance involves other tissues and if prolonged exposure to TH in adult life could have a similar effect. This is currently under investigation, as prolonged suppression of TSH was for many years advocated as standard treatment of differentiated thyroid cancer (16), and the mechanism of refractoriness to TH in some patients without THRB gene defects remains unresolved (17,18).
Footnotes
Acknowledgments
We are grateful to the individuals who consented to participate in this study. This work was supported in part by grants DK 15070 to S.R. and DK110322 to A.M.D. from the National Institutes of Health (Bethesda, MD).
Author Disclosure Statement
No competing financial interests exist.