
Abstract
Hürthle cell tumors (HCT), including Hürthle cell adenomas (HCA) and Hürthle cell carcinomas (HCCs), arise in the thyroid gland and are defined in part by an accumulation of mitochondria. These neoplasms were long considered a subtype of follicular neoplasm, although HCT is now generally considered a distinct entity. HCTs exhibit overlapping but distinct clinical features compared to follicular tumors, and several studies have demonstrated that HCTs harbor distinct genomic alterations compared to other forms of thyroid cancer. Two studies recently reported the most complete characterization of the HCC genome to date. These studies assessed complementary cohorts of HCC specimens. The study by Ganly et al. consisted of a large panel of primary HCCs, including 32 widely invasive and 24 minimally invasive primary tumors. Exome and RNA sequencing of material isolated from fresh-frozen tumor specimens was performed. The study by Gopal et al. utilized exome and targeted sequencing to characterize the nuclear and mitochondrial genomes of 32 primary tumors and 38 resected regional and distant metastases using DNA isolated from formalin-fixed paraffin-embedded tissues. Here, HCC is briefly reviewed in the context of these studies.
Clinicopathologic Features of Hürthle Cell Tumors
Hürthle cell carcinoma (HCC) is a rare type of well-differentiated thyroid cancer, accounting for approximately 5% of thyroid cancer diagnoses. These tumors are composed of Hürthle cells that contain an abundance of abnormal mitochondria and prominent nucleoli, and exhibit a loss of cell polarity. The Hürthle cell itself is believed to derive from the follicular epithelial cell. These cells are present in benign thyroid conditions, including benign nodular goiter and chronic lymphocytic (Hashimoto's) thyroiditis. However, in Hürthle cell tumors (HCTs), Hürthle cells comprise >75% of the tumor volume (1).
The Hürthle cell was first described by Max Askanazy, a German-Swiss pathologist, in 1898, but was mistakenly named after the German physiologist Hürthle who actually described the parafollicular C cell. The first description of HCC was published by Theodor Langhans in 1907 (2). The abundance of mitochondria imparts an intensely eosinophilic granular cytoplasm. Hürthle cells are also called oncocytic cells and develop in other tissues, including the salivary gland, kidney, pituitary gland, and parathyroid gland. Similar to the thyroid gland, oncocytic cells in other tissues may accumulate and give rise to neoplasms such as Warthin's tumor in the parotid gland and renal oncocytoma in the kidney.
In the thyroid gland, both Hürthle cell adenomas (HCA) and HCCs can develop. The differentiation between adenoma and carcinoma is determined by the presence of capsular and/or vascular invasion (3). HCCs are further categorized according to the extent of invasion. Minimally invasive HCCs demonstrate only capsular invasion or present with only a few foci (<4) of vascular invasion. Widely invasive HCCs exhibit extensive capsular invasion, extrathyroidal extension, and/or more than four foci of vascular invasion. These features are illustrated in Figure 1. The distinction between adenoma, minimally invasive HCC, and widely invasive HCC is crucial because this predicts clinical outcome. HCAs are benign and cured by thyroid lobectomy. Minimally invasive HCCs also exhibit an indolent course and very rarely metastasize to lymph nodes or distant sites. In contrast, widely invasive HCCs are more locally invasive, and they often metastasize to regional lymph nodes and distant metastatic sites, including the lung and bone. Figure 2 illustrates a patient with widely invasive HCC. This patient had a large tumor invading the overlying strap muscles and laryngopharynx, as shown by contrast-enhanced computed tomography. The tumor also metastasized to the lung, with multiple 18F-fluorodeoxyglucose–avid pulmonary nodules. In a review of 59 patients by Stojadinovic et al., a rate of distant metastases of 33% and a rate of nodal metastasis of 21% were reported (4). Ruegemer et al. also demonstrated a rate of distant metastases of 34% (5).

Hürthle cell carcinoma (HCC; widely invasive) showing vascular invasion.
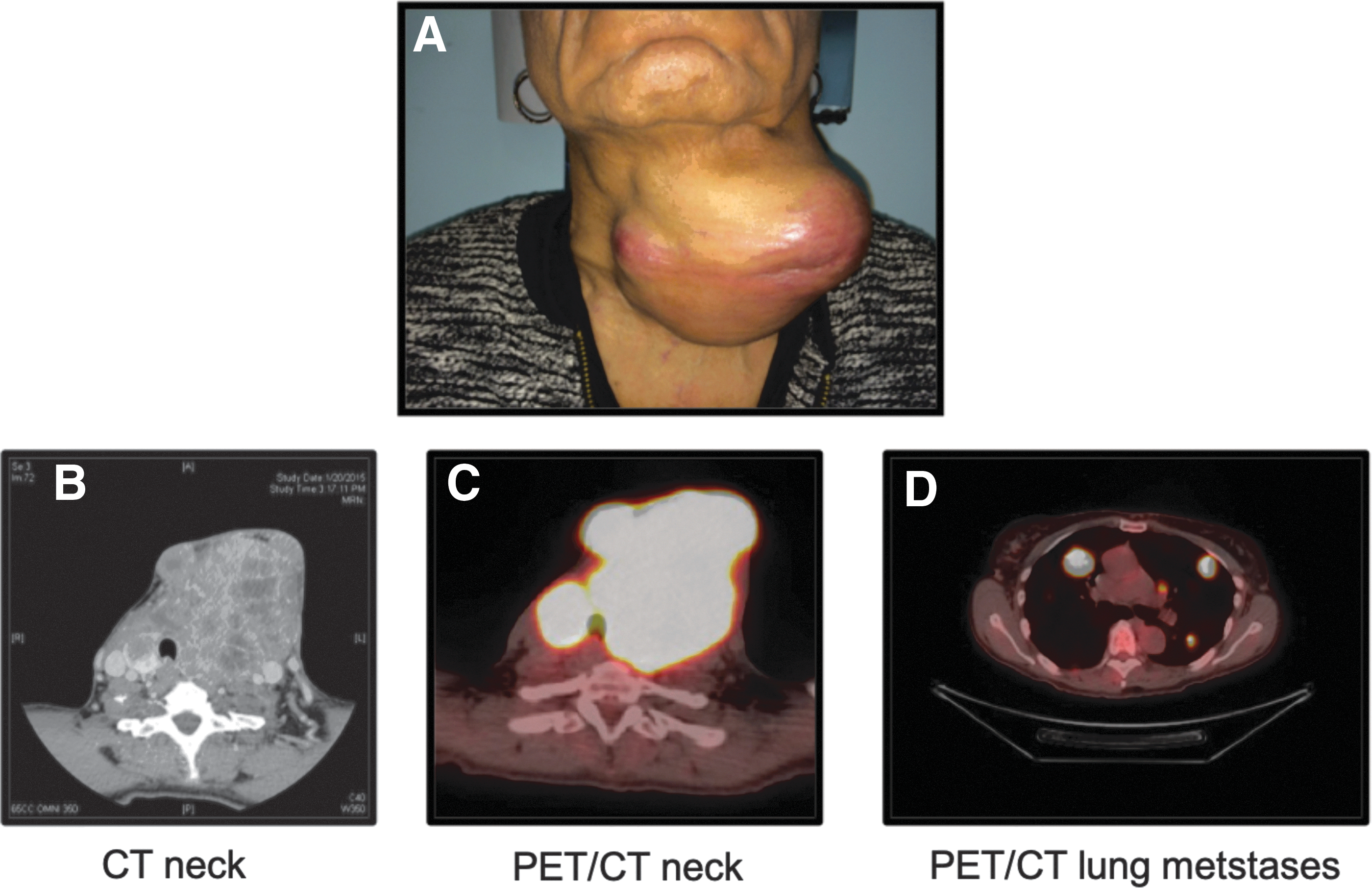
Clinical presentation of patient with widely invasive HCC. (
HCCs are typically radioactive iodine refractory and respond poorly to chemotherapy and radiation (6,7). Outcomes for patients with widely invasive HCC are poorer compared to patients with minimally invasive HCC (3,8). In a study of 33 patients treated over 25 years at the University of California at San Francisco, Kushchayeva et al. reported a rate of metastasis of 36%, a recurrence rate of 24%, and disease-specific survival of 74% and 49% at 5 and 10 years respectively (9). Lopez-Penabad et al. reported a similarly high mortality rate of 40% associated with HCC in 89 patients managed at the MD Anderson Cancer Center over a period of 50 years (8). A Surveillance, Epidemiology, and End Results study by Bhattacharyya also reported on 555 cases of HCC from 1988 to 1998, showing overall mean 5- and 10-year survival rates of 85% and 71%, respectively (10).
Nuclear Mutations in HCC
Previous DNA sequencing studies of HCC reported a different mutational profile in HCC compared to papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC). In PTC, the predominant genetic alterations include activating mutations of the BRAF gene occurring in 40–49% of patients with classical PTC (11 –13). However, the BRAFV600E mutation rarely occurs, if at all, in HCC (14). RAS mutations, which activate the RAS-RAF-MEK pathway, were the next most frequently identified mutation in well-differentiated thyroid cancer. RAS family mutations (NRAS, KRAS, and HRAS) were reported to occur in 45% of follicular tumors, whereas these mutations were uncommon (∼10%) in PTC. Previous studies of HCC reported an incidence of RAS mutations of 10% (14,15). Other mutations observed in FTC, including PIK3CA (10–30%), were also rare in HCTs (14,15).
Ganly et al. (16) observed a higher mutational burden in HCC compared to PTC. HCCs harbored an average of 2.6 mutations/Mb, which was sixfold greater than reported in The Cancer Genome Atlas (TCGA) of PTC (0.4 mutations/Mb). This mutational burden was comparable to glioblastoma multiforme, cervical cancer, and ovarian cancer. In contrast, Gopal et al. (17) reported a mutation frequency in minimally invasive HCC to be similar to PTC (0.4 non-silent mutations/Mb). Resected metastases harbored a higher mutation frequency of 0.7 mutations/Mb. The reasons underlying the difference in mutational burden reported by the two studies remain unclear, but differences between material isolated from fresh-frozen and formalin-fixed paraffin-embedded samples or the data analysis methods might contribute. Nonetheless, Ganly et al. (16) identified mutations impacting the RAS/RAF/MAPK and PI3K/AKT/MTOR pathways in 55% of tumors (Fig. 3A). Mutations were identified in receptor tyrosine kinases (20%), PI3-kinase mutations (2%), PTEN mutations (4%), TSC1/2 mutations (7%), and NF1 gene deletions or mutations (9%). Mutations in NRAS, HRAS, or KRAS occurred in 15% of tumors (NRAS 9%, HRAS 2%, and KRAS 4%). Mutations in genes involved in translation initiation downstream of RAS/RAF/MAPK and mTOR were also identified. These included EIF1AX mutations (11%) and mutations in other EIF1,2,3 genes (8%). Mutations in EIF1AX were previously reported in uveal melanoma and in approximately 1% of PTC (11,18). EIF1AX mutations have also been identified in other clinically aggressive forms of thyroid cancer, including 11% of poorly differentiated thyroid cancers and 9% of anaplastic thyroid cancers (19).
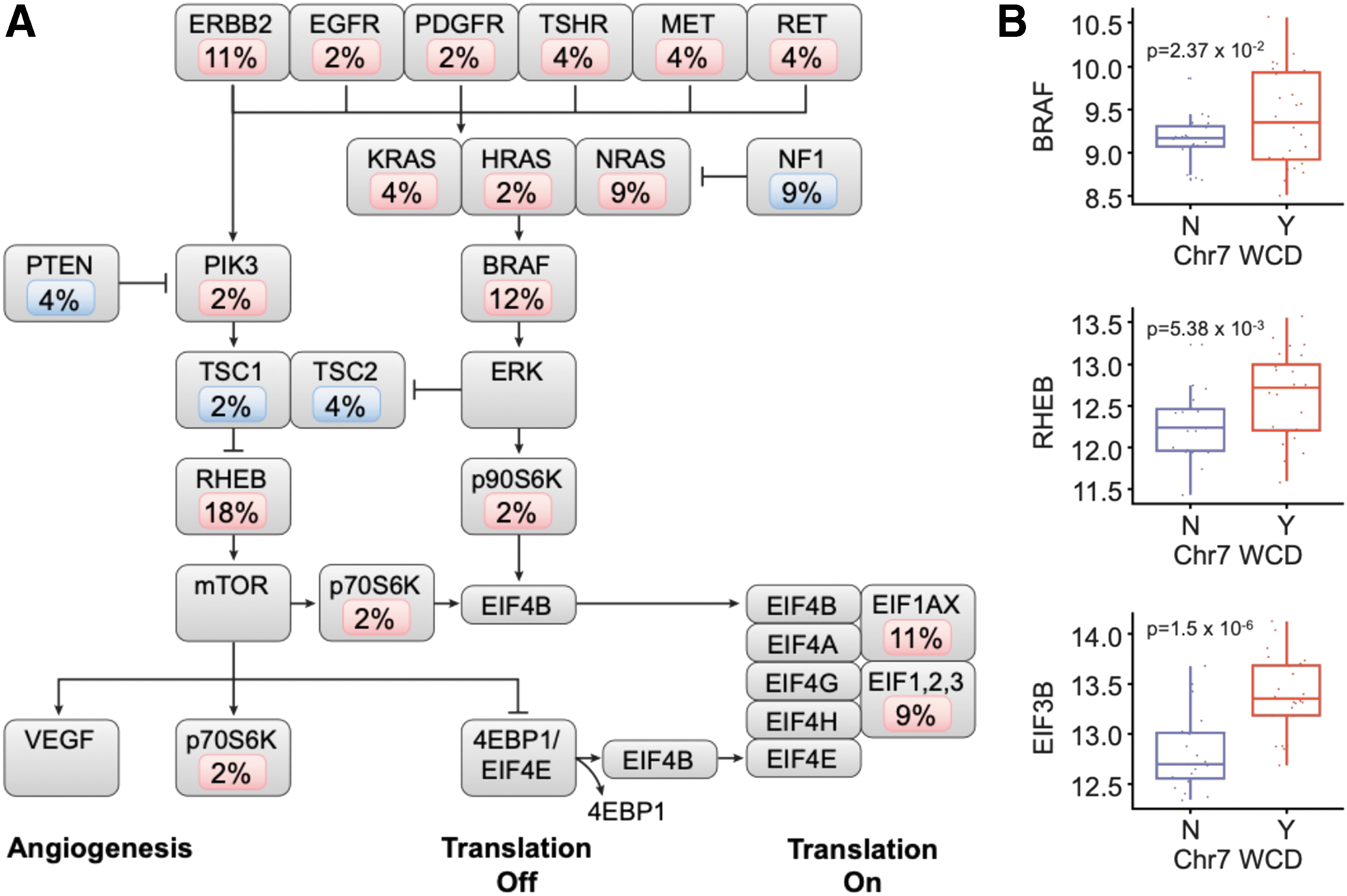
Reproduced with permission from Ganly et al. (16). Somatic mutations of the RAS/RAF/MAPK and PI3K/AKT/mTOR pathways. (
In addition to point mutations, amplification of chromosomes 5 and 7 was observed and was correlated with an increased abundance of transcripts of several genes in the RAS/RAF/MAPK and PI3K/AKT/MTOR pathways encoded on chromosomes 5 and 7. For example, tumors with amplification of chromosome 7 had a statistically significant overexpression of several genes involved in mTOR signaling and protein translation (Fig. 3B). These genes included BRAF (p = 0.0237), RHEB (p = 0.005), and EIB3B (p = 1.5 × 10–6). The eukaryotic initiation factor 3 (EIF3) complex is regulated by mTOR signaling and is essential for the initiation of protein synthesis (20). Elevated EIF3B expression has been shown in several cancers, including human bladder cancer and prostate cancer, esophageal cancer, and glioblastoma (21 –23).
In addition to RAS/RAF/MAPK and PI3K/AKT/MTOR pathway alterations, mutations in the DNA damage/repair pathway were identified (Fig. 4A). These included mutations in the TP53 tumor suppressor gene (7%) and ATM (5%). Mutations in the DNA repair pathways of homologous recombination, nucleotide excision repair, mismatch repair, and DNA strand crosslink repair were also identified, including mutations in the nucleotide excision repair gene ERCC5 (13% of tumors).
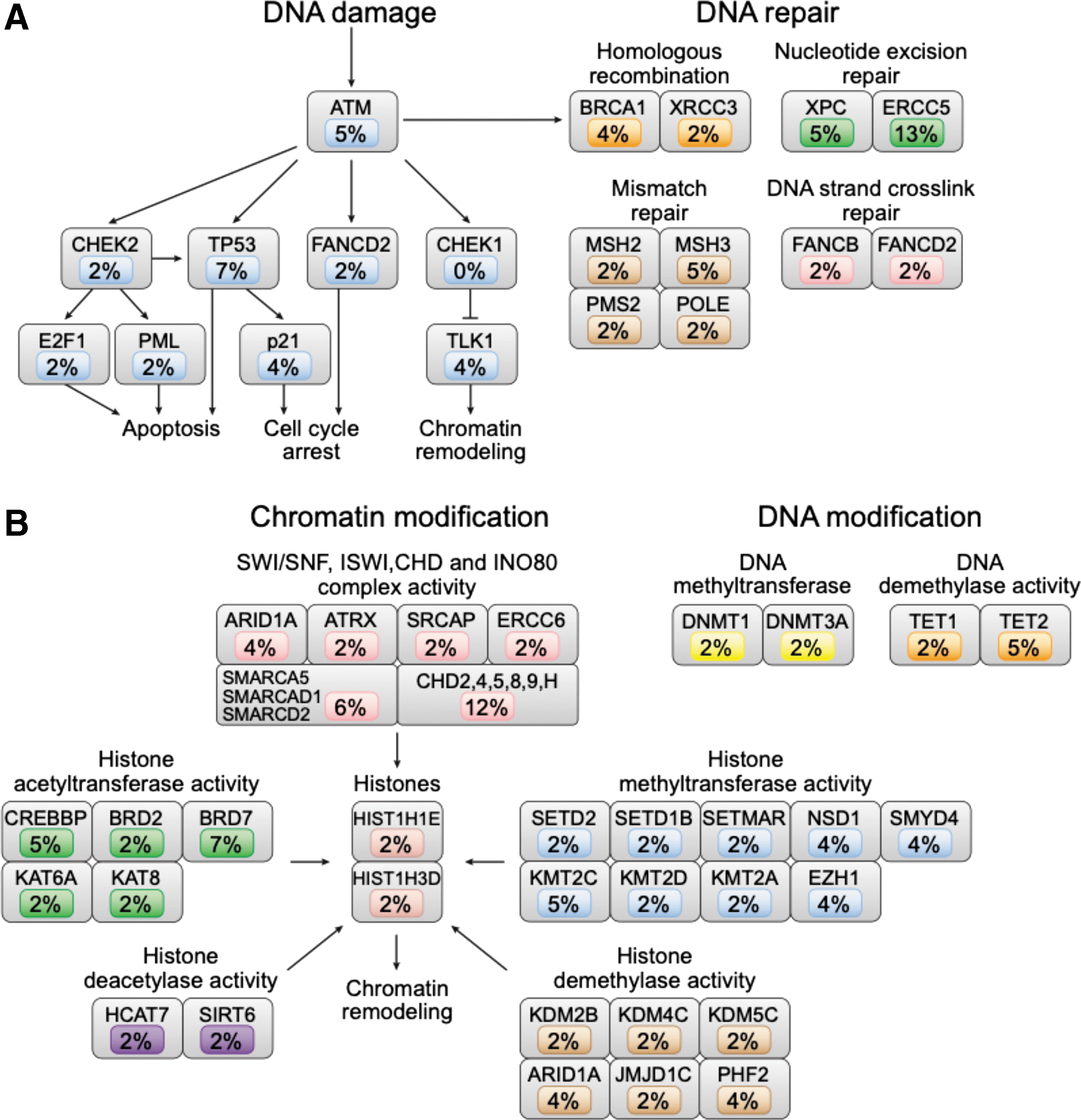
Reproduced with permission from Ganly et al. (16). Somatic mutations of the canonical signal transduction and tumor suppressor pathways in HCC. (
Ganly et al. (16) also reported mutations in genes encoding epigenetic modification enzymes in 60% of HCCs, including components of the SWI/SNF, ISWI, CHD, and INO80 complexes (Fig. 4B). Consistent with shared function of epigenetic modifiers in HCC, mutations in these genes were mutually exclusive with one another.
Mutations in the TERT promoter were also identified by both studies. TERT promoter mutations have been associated with clinically aggressive forms of thyroid cancer (24). Consistent with this association, Ganly et al. (16) reported TERT promoter mutations in 32% in widely invasive HCCs and only 5% in minimally invasive HCCs (Fig. 4C). Alterations in alternate mechanisms of telomere elongation were also identified, including mutations in DAXX and ATRX. These mutations were mutually exclusive with TERT promoter mutations, suggesting multiple events impinge upon telomere lengthening in clinically aggressive HCC.
Gene Rearrangements
Chromosomal translocations involving the RET proto-oncogene have been recurrently observed in PTC. In FTC, PAX8/PPAR-γ rearrangements have been reported in 25–60% of tumors (25). Rearrangements leading to other oncogenic fusions were also identified in PTC in TCGA study involving BRAF, THADA, ALK (EML4/ALK), and NTRK3 (ETV6/NTRK3 and RBPMS/NTRK3) (11). These fusions were infrequently observed in HCC in the current studies. Using transcriptome sequencing, however, Ganly et al. (16) identified several novel fusion genes in HCC. These included recurrent in-frame coding rearrangements of CHCHD10_VPREB3 (chromosome 22), HEPHL1_PANX1 (chromosome 11), TMEM233_PRKAB1 (chromosome 12), ACSS1_APMAP (chromosome 20), RSPH6A_DMWD (chromosome 19), DUOXA1_DUOX2 (chromosome 15), OSGIN1_NECAB2 (chromosome 16), BCAP29_SLC26A4 (chromosome 7), and TFG_GPR128 (chromosome 3). Functional studies will be required to determine whether these fusions act as driver events in HCC.
Widespread Chromosomal Losses are a Hallmark of HCC
Corver et al. first reported widespread loss of chromosomes in a series of HCCs, and Kasain et al. found evidence for widespread chromosome loss in two HCC studies using whole-genome sequencing (26,27). The studies of Ganly et al. (16) and Gopal et al. (17) also observed widespread chromosomal loss as a common feature of HCC in these larger tumor cohorts (Fig. 5).
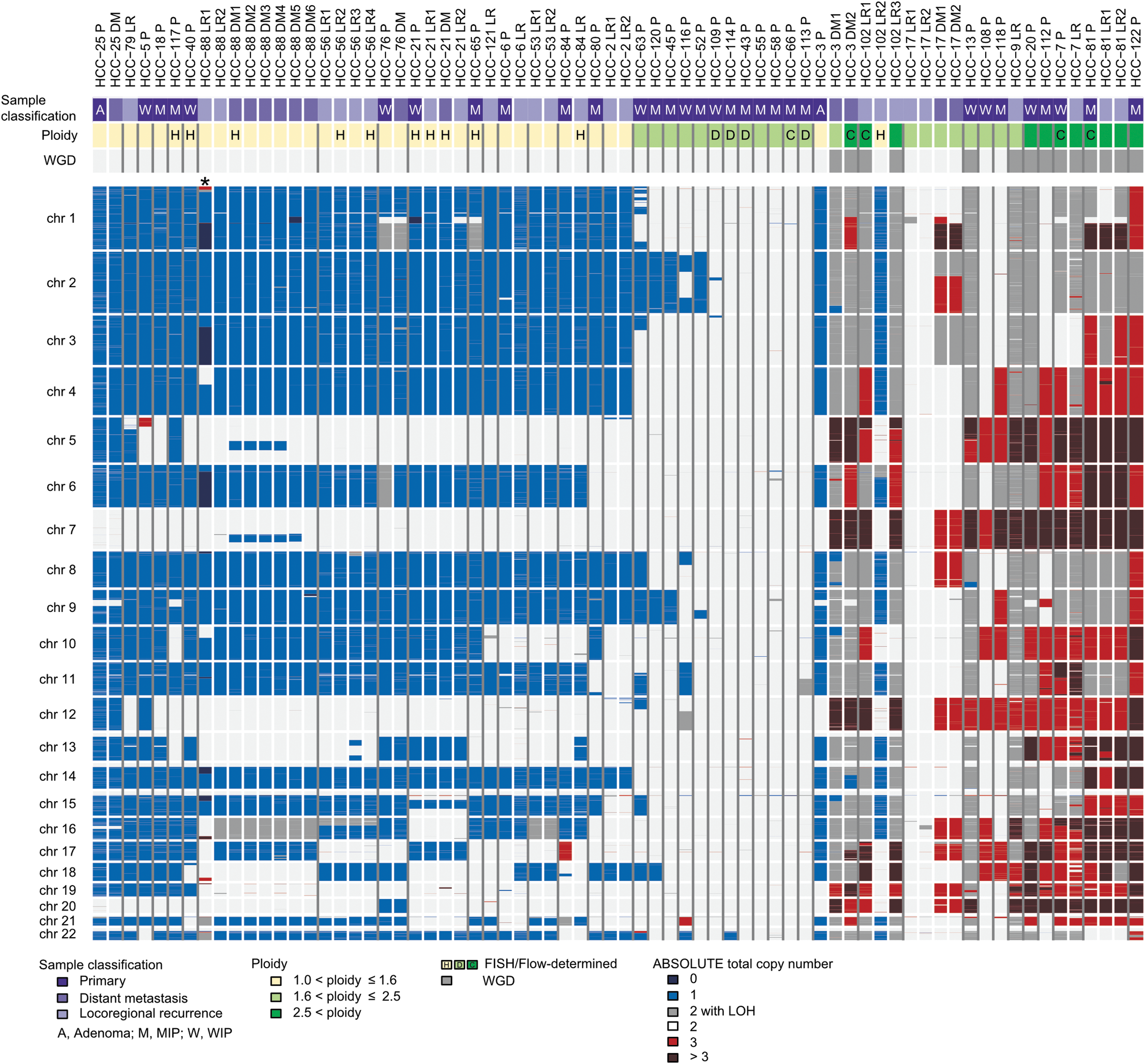
Widespread loss of chromosomes in HCC. Reproduced with permission from Gopal et al. (17). Absolute total copy numbers of individual segments are delineated by their genomic position along the 22 chromosomes (top to bottom). Sample identifiers and classifications are shown at the top. Patients are ordered according to mean ploidy of “aggregated tumors,” and individual tumor samples from the same patient are ordered based on the date of resection. The presence of whole-genome doubling is indicated. Ploidy results of fluorescence in situ hybridization and imaging flow cytometry (flow) are indicated by letters. Colors indicate the loss, copy neutral loss of heterozygosity, or gain at genomic loci. *HCTC-88 LR1 analysis was confounded by low tumor purity; although several chromosomes are predicted to be completely lost, these data more likely reflect single copy loss in tumor cells or a tumor subclone.
These studies provide support to a model of genome evolution in which HCCs appear to evolve through a near-haploid state in which one copy of most chromosomes is lost (26). Whether chromosome loss is progressive or occurs as a consequence of a “catastrophic” event is not established. However, it is notable that several tumors reported by Gopal et al. (17) harbored single losses of chromosomes 2, 9, 11, and 18, which could suggest these are early events in this process. Chromosomes 12, 20, and 5 were infrequently lost. Chromosome 7 was never lost and, further, was frequently amplified in HCC. As described above, Ganly et al. (16) reported that chromosome 7 amplification was associated with increased transcription of genes involved in RAS/RAF/MAPK and PI3K/AKT/mTOR signaling. Chromosome 7 is also heavily imprinted, raising another mechanism by which its loss might be selected against (28).
In some tumors, the near-haploid genome underwent a whole-genome duplication event, restoring a near-diploid or a polysomic chromosome number. Ganly et al. (16) reported that whole-genome duplication leading to a complex or polysomic genotype was observed more frequently in widely invasive HCC. Even though two copies of the haploid chromosomes were restored, the duplicated copies arose from a single parent chromosome—a term called “uniparental disomy.” This resulted in widespread loss of heterozygosity (LOH) in HCC.
In both studies, widespread LOH was associated with a poorer prognosis, suggesting that widespread chromosomal losses contributed to more aggressive disease. The selective advantage underlying widespread loss of chromosomes is not known. In the near-haploid state, tumor suppressor gene inactivation can be theoretically achieved by a single inactivating mutation in contrast to the “two-hit” process required to inactivate tumor suppressors in the context of a diploid genome. Consistent with this notion, Gopal et al. (17) observed mutations in tumor suppressor genes to be more common in tumors with widespread LOH.
By comparing the genomes of primary and metastatic tumors resected from the same patients, the study by Gopal et al. (17) retraced the evolution of chromosome content during metastatic progression. Unlike most other forms of cancers in which widespread loss of chromosomes has been observed, the near-haploid state was frequently maintained in metastatic tumors that arose from a near-haploid primary tumor. Therefore, maintenance of near-haploid DNA content was not inherently disadvantageous to metastatic disease progression. In one patient, however, a near-haploid primary tumor underwent genome duplication, leading to distant metastases harboring polysomic genomes.
Chromosomal losses were previously reported not to be associated with mtDNA mutations (see below), and the mechanisms underlying chromosome loss have not been established (29). However, one group has proposed that dysfunctional mitochondria contribute to activation of DNA damage pathways, which leads to chromosomal missegregation (30).
Gene Expression and Pathway Analysis of HCC
Ganly et al. (16) performed transcriptome sequencing in order to identify expression profiles associated with clinical outcomes. Unsupervised clustering of the gene expression profiles confirmed that widely invasive HCC associated with alterations in EIF2 signaling, EIF4 and p70S6K signaling, and mTOR signaling. In addition, alterations in mitochondrial dysfunction and oxidative phosphorylation were associated with widely invasive HCC. Integration of transcriptomic and mutational data identified three subtypes of HCC. The first subtype was enriched with widely invasive HCC and TERT alterations, widespread LOH, and chromosome 7 amplification. The majority of recurrences and deaths occurred in this group. The second subtype was enriched in tumors without TERT alterations but harbored widespread LOH and chromosome 7 amplifications. This group exhibited a lower frequency of recurrence and death. The third subtype was enriched in minimally invasive HCC without TERT alterations, LOH, or chromosome 7 amplification. There were no recurrences or deaths observed in this group. If validated on additional cohorts of patients, this integration of genomic and transcription data might aid in the identification of patients at high risk for recurrence and death and impact treatment decisions for these patients.
Recurrent Mitochondrial Mutations in HCC
Mitochondrial accumulation is a defining feature of oncocytic neoplasms, including HCC. Several studies have examined mitochondrial function in HCC using the XTC.UC1 tumor cell line generated from a mammary gland metastasis of HCC (31). Differing activities of complex I have been reported in XTC.UC1 cells (32 –34). Mutations in complex I (ND1) and complex III (CYTB) were identified in XTC.UC1, prompting the authors to search for mitochondrial mutations in a panel of oncocytic thyroid and breast neoplasms (32 –34). Gasparre et al. indeed reported that 25/45 (56%) oncocytic thyroid neoplasms harbored mutations in complex I encoding genes (35).
Gopal et al. (17) and Ganly et al. (16) extended mtDNA analysis to include a larger panel of HCCs, compared HCCs to large studies of mtDNA variants observed across cancer types, and provided quantitative estimates of the fraction of mitochondrial genomes harboring mutations. In addition, Gopal et al. (17) further analyzed mtDNA mutations in primary tumors and resected metastases from individual patients (Fig. 6).
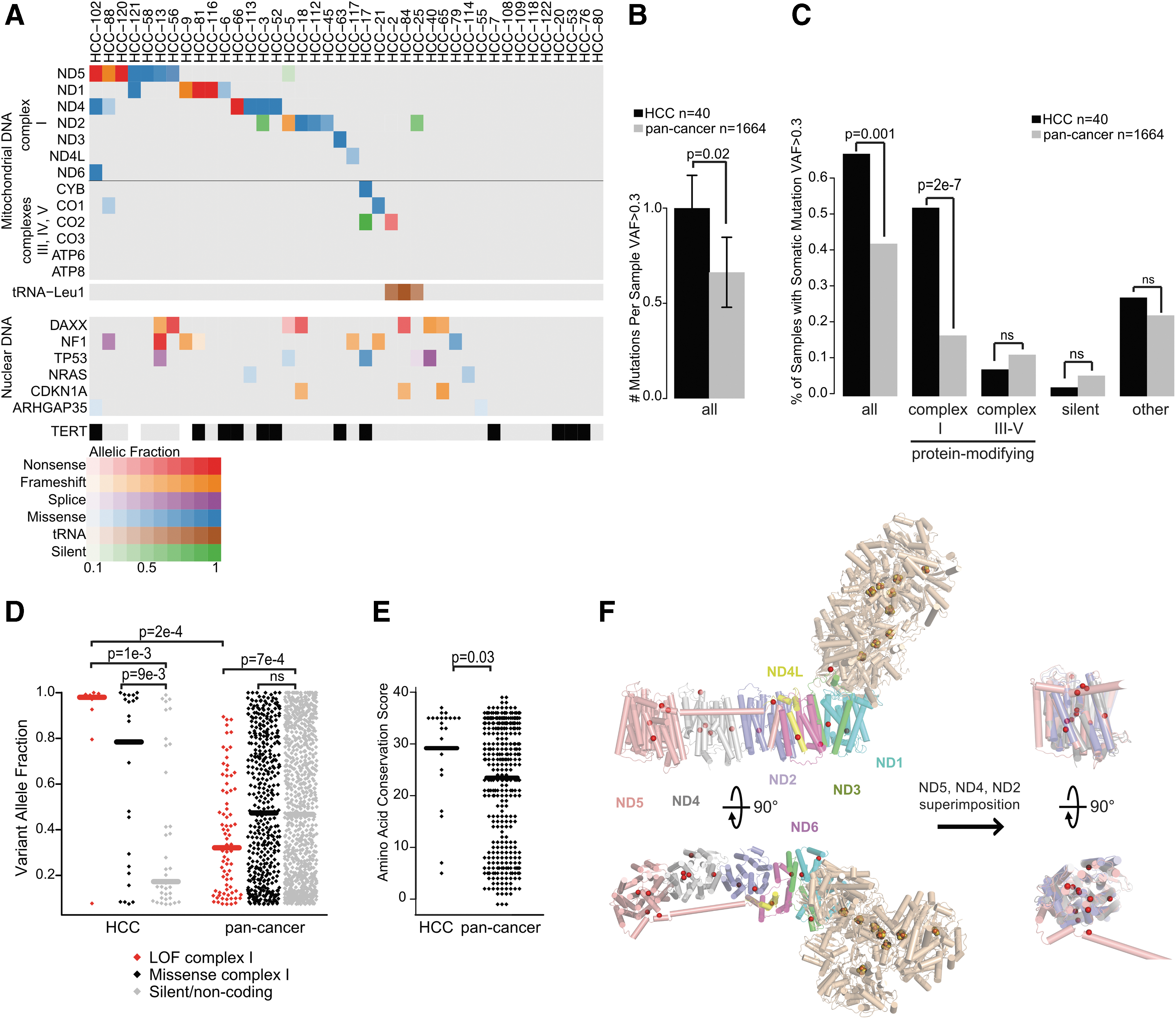
Mitochondrial DNA mutations in HCC. Reproduced with permission from Gopal et al. (17). (
Three important findings relating to mtDNA mutations emerged from these studies. First, consistent with the study by Gasparre et al. (34), mtDNA mutations were specifically enriched in genes encoding components of complex I. Second, mtDNA mutations, and specifically disruptive mutations in complex I, were more frequent in HCC compared to other cancers (36). Third, when present, disruptive mtDNA mutations existed in the vast majority of mitochondria within HCCs. Finally, disruptive complex I mutations were maintained in metastases when the primary tumor harbored the mtDNA mutation. Together, these findings support the hypothesis that disruptive complex I mutations were under positive selection, and therefore acted as cancer-driving events in HCC. However, recurrent nuclear-encoded mutations in components of complex I were not identified in these studies, including mutations in NDUFA13 (GRIM-19) that had been previously reported (37,38). Also, mtDNA mutations were not found in all HCCs. Therefore, additional mechanisms beyond mtDNA mutations might underlie the accumulation of mitochondria and metabolic reprogramming in HCC.
The origin and function of mtDNA mutations in HCC are not well established. Although mtDNA mutations have been observed in oncocytic metaplasia associated with autoimmune (Hashimoto's) thyroiditis, the events observed in HCC are not the same as those reported in autoimmune thyroiditis (39). Further, the biochemical consequence of complex I mutations have not been elucidated. Therefore, it will be important to determine whether mtDNA mutations observed in HCC impair complex I function, and to determine whether mtDNA mutations promote the growth of HCC.
Relationship of HCC to Other Cancers
The studies of Ganly et al. (16) and Gopal et al. (17) support the recent classification of HCC as a distinct form of thyroid cancer. These tumors harbor distinct genomes compared to FTC and PTC, as described above. In particular, HCCs harbor fewer mutations in canonical FTC driver genes, including RAS and PAX8/PPAR-γ. mtDNA-encoded mutations in components of complex I and widespread loss of chromosomes appear to represent the dominant genetic features of HCC.
Although mtDNA mutations are considered disadvantageous to most cancers, oncocytic tumors arising in different tissues also harbor a higher frequency of mtDNA alterations, including events predicted to impair complex I function (35,36,40,41). These mtDNA mutations have been proposed to be a barrier to malignant transformation, in part because most oncocytic neoplasms exhibit benign prognoses. However, the presence of disruptive homoplasmic (i.e., mitochondria with identical DNA) mtDNA mutations in metastatic HCC suggests that these events are not barriers to transformation or tumor progression in some contexts.
Adrenocortical carcinoma, germ-cell tumors, chromophobe renal-cell carcinoma, and most recently mesothelioma have also been reported to harbor widespread chromosomal losses and LOH (40,42 –44). Therefore, understanding the origin and consequences of widespread loss of chromosomes is likely to have implications beyond HCC.
Conclusion
The studies by Gopal et al. (17) and Ganly et al. (16) support the notion that HCC is a distinct form of thyroid cancer. Mechanistic studies should now be pursued to determine the origin and selective advantage of widespread chromosomal losses and mtDNA mutations, and the series of novel putative driver mutations identified by these studies. The study by Ganly et al. also provides an important rationale for the ongoing assessment of small molecule inhibitors of the mTOR pathway in HCC, which have demonstrated promising early response rates (45).
HCC represents an “outlier” form of cancer, harboring a high frequency of mtDNA mutations and whole chromosome losses. These events occur at a lower frequency across many types of cancer. HCC therefore represents an important cancer through which the origin and function of these alterations might be elucidated.
Footnotes
Author Disclosure Statement
No competing financial interests exist.