
Abstract
Background:
A novel form of thyroid hormone (TH) signaling is represented by 3-iodothyronamine (T1AM), an endogenous TH derivative that interacts with specific molecular targets, including trace amine-associated receptor 1 (TAAR1), and induces pro-learning and anti-amnestic effects in mice. Dysregulation of TH signaling has long been hypothesized to play a role in Alzheimer's disease (AD). In the present investigation, we explored the neuroprotective role of T1AM in beta amyloid (Aβ)-induced synaptic and behavioral impairment, focusing on the entorhinal cortex (EC), an area that is affected early by AD pathology.
Methods:
Field potentials were evoked in EC layer II, and long-term potentiation (LTP) was elicited by high frequency stimulation (HFS). T1AM (5 μM) and/or Aβ(1-42) (200 nM), were administered for 10 minutes, starting 5 minutes before HFS. Selective TAAR1 agonist RO5166017 (250 nM) and TAAR1 antagonist EPPTB (5 nM) were also used. The electrophysiological experiments were repeated in EC-slices taken from a mouse model of AD (mutant human amyloid precursor protein [mhAPP], J20 line). We also assessed the in vivo effects of T1AM on EC-dependent associative memory deficits, which were detected in mhAPP mice by behavioral evaluations based on the novel-object recognition paradigm. TAAR1 expression was determined by Western blot, whereas T1AM and its metabolite 3-iodothyroacetic acid (TA1) were assayed by high-performance liquid chromatography coupled to mass spectrometry.
Results:
We demonstrate the presence of endogenous T1AM and TAAR1 in the EC of wild-type and mhAPP mice. Exposure to Aβ(1–42) inhibited LTP, and T1AM perfusion (at a concentration of 5 μM, leading to an actual concentration in the perfusion buffer ranging from 44 to 298 nM) restored it, whereas equimolar amounts of 3,5,3′-triiodo-L-thyronine (T3) and TA1 were ineffective. The response to T1AM was abolished by the TAAR1 antagonist EPPTB, whereas it was mimicked by the TAAR1 agonist RO5166017. In the EC of APPJ20 mice, LTP could not be elicited, but it was rescued by T1AM. The intra-cerebro-ventricular administration of T1AM (0.89 μg/kg) also restored recognition memory that was impaired in mhAPP mice.
Conclusions:
Our results suggest that T1AM and TAAR1 are part of an endogenous system that can be modulated to prevent synaptic and behavioral deficits associated with Aβ-related toxicity.
Introduction
Thyroid hormones (TH) have an established role in the development of the central nervous system (1), and they may also play a role in dementia and in Alzheimer's disease (AD) (2 –7). Animal studies have supported a role of TH as neuroprotective agents in brain areas that are affected early in AD. Indeed, TH administration was shown to reduce hippocampal neuronal damage induced by ischemia and to protect neurons from glutamate-induced death (8,9). However, hypothyroidism reduced hippocampal neurogenesis (10) and in CA1 neurons, it impaired long-term potentiation (LTP), which was then restored by TH administration (11,12). Moreover, TH has been demonstrated to influence amyloid precursor protein (APP) transcription, the alternative splicing of APP mRNA, and APP protein processing (13 –15) and to rescue memory deficits in AD rodent models (16 –19). These results suggest that TH may exert a neuroprotective role against beta amyloid (Aβ)-dependent neuronal impairment, which is assumed to be one of the pathophysiological mechanisms involved in AD.
In recent years, the picture of TH signaling has become more complex than originally believed. The canonical concept that TH receptors behave as ligand-dependent transcription factors is well established, but the relevance of noncanonical actions has also been recognized (20). Another significant breakthrough has been the discovery of novel TH derivatives acting on receptors that differ from nuclear TH ones. In particular, 3-iodothyronamine (T1AM), allegedly derived from TH through decarboxylation and deiodination, has been reported to be a chemical messenger that activates a G protein-coupled receptor known as trace amine-associated receptor 1 (TAAR1) with high potency (21). TAAR1 is widely expressed in the brain (22,23); it has been implicated in several neuropsychiatric disorders and has attracted attention for being a potential drug target (24 –26). The intra-cerebro-ventricular (i.c.v.) administration of T1AM in mice produced a pro-learning and anti-amnestic response (27,28). Further, it has been suggested that some of T1AM effects could be due to its oxidative product, 3-iodothyroacetic acid (TA1) (28 –30).
In this work, we aimed at determining whether T1AM may play a protective role in a specific model of neuronal injury, namely Aβ-dependent synaptic and behavioral impairment. We focused our study on the layer II of the entorhinal cortex (EC), an area that is affected early in AD (31,32). The rationale for this choice is also related to previous observations showing that EC horizontal connections are vulnerable to the effects of exogenously applied Aβ(1 –42) oligomers (33,34). Moreover, in previously published projects, we showed that EC synaptic function is affected early in mutant human APP (mhAPP) transgenic mice (J20 line), and that the synaptic dysfunction is associated with an impairment of specific forms of associative memory (35), which depend on EC functional integrity (36,37).
Our results show that exogenous T1AM is able to rescue the EC in both models of Aβ-toxicity. This finding adds a novel issue to the discussions on the elusive links between TH signaling, Aβ effects, and the pathophysiology of AD.
Materials and Methods
Animals
Transgenic mhAPP mice (APPsweInd, line J20) overexpressing an alternatively spliced human APP minigene that encodes hAPP695, hAPP751, and hAPP770, bearing mutations linked to familial AD (38), were used, together with their littermate controls (C57BL/6J). All experiments were conducted in male mice at the age of two months, in accordance with the Italian Ministry of Health and the European Community guidelines (Legislative Decree no. 116/92 and European Directive 86/609/EEC). The experimental protocol (IACUC document) was approved by the Ministry of Health (protocol no. 192/2000-A). Two-month-old TAAR1 knockout (KO) mice (C57BL/6J × 129 Sv/J) and wild-type (WT) littermates were generously provided by Stefano Espinoza (Istituto Italiano di Tecnologia, Genova, Italy).
Drugs
T1AM and TA1 were synthesized as previously described (39) and were generously provided by Dr. Thomas Scanlan (Oregon Health & Science University); 3,5,3′-triiodo-L-thyronine (T3) and TAAR1 antagonist EPPTB were purchased from Sigma-Aldrich (St. Louis, MO); and the TAAR1 agonist RO5166017 was generously provided by Dr. Raul Gainetdinov (University of St. Petersburg). Aβ(1 –42) was purchased from Abbiotec. Oligomeric Aβ(1 –42) peptide was prepared as previously described and characterized by atomic force microscopy (40) and mass spectrometry (41). Aliquots were stored at −20°C in dimethyl sulfoxide as a 200 mM stock solution and diluted to the desired final concentration in artificial CSF (ACSF), containing the following (in mM): 119 NaCl, 2.5 KCl, 2 CaCl2, 1.2 MgSO4, 1 NaH2PO4, 6.2 NaHCO3, 10 HEPES, and 11 glucose.
In vitro electrophysiology
Electrophysiology was performed as in Origlia et al. (33,34,41). Briefly, mice were anesthetized with urethane i.p. injections (20% sol., 0.1 mL/100 g body weight) and then decapitated. Horizontal EC-hippocampal slices were produced by using a vibratome (Leica VT1200S). All steps were performed in ice-cold oxygenated ACSF. Slices were then transferred to a chamber and perfused at a 2–3 mL/min rate. Field potentials (FPs) were evoked by a concentric bipolar stimulating electrode in the layer II of EC. Basal recording was carried out by using stimulus intensity evoking a response whose amplitude was 50–60% of the maximal amplitude. After 15 minutes of stable baseline, LTP was induced by high frequency stimulation (HFS, three trains of 100 pulses at 100 Hz, 10 seconds interval). After HFS, FPs were monitored every 20 seconds for at least 40 minutes. The magnitude of LTP was calculated as the average of the relative amplitudes (compared with baseline) of FPs recorded in the last 10 minutes. Values were expressed as percentage change relative to the baseline. Data collection and analysis were performed blindly by two different operators. Aβ (200 nM), T1AM (5 μM), TAAR1 antagonist EPPTB (5 nM), and TAAR1 agonist RO5166017 (250 nM) were added to ACSF perfusion and administered for 10 minutes, 5 minutes before and 5 minutes after HFS.
I.c.v. injection
Drug administration was performed under avertin-induced anesthesia according to the method described (27,42), with minor modifications. The depth of anesthesia was checked by monitoring the respiratory rate (reduced within two minutes) and testing for lack of a pain response to gentle pressure on the hind paws. The head of the anesthetized mouse was positioned into a stereotaxic system and firmly blocked. A fine needle was inserted perpendicularly through the skull into the brain at the coordinates of one of the lateral ventricles, identified according to the atlas of mouse brain (y: −0.3 mm/x: −1 mm/z: −1.5 mm). Ten microliters were then slowly injected (in 20 seconds) into a lateral ventricle as per the protocol described by Manni et al. (27). Immediately after needle removal, the animal remained quiet for approximately one minute and then resumed its normal activity.
Behavioral study
Behavioral testing was performed as described (37). To habituate the mice to the experimenter, they underwent extensive handling for one week before the experiments. Behavioral testing proceeded in two stages: habituation and performance of specific tasks that included novel object-place recognition task (in OPRT) and novel object-place-context recognition task (in OPCRT), as described in Wilson et al. (37). Habituation and testing took place in a 60-cm square box (arena) with 40-cm high walls that could be shaped with two sets of contextual features (to be used in the OPCRT). The objects were easily cleanable household objects approximately the same size as a mouse in at least one dimension, and made from plastic, metal, or glass. During the habituation phase, the mice were exposed to the arena for one hour for three consecutive days before the test. Vehicle or T1AM was injected i.c.v. on the third day, then OPRT and OPRCT took place one hour after the recovery from i.c.v. injections.
Object exploration was monitored via an overhead camera. Exploration time was defined as the time spent with the object in close proximity (within 2 cm), with the nose directed toward it, or sniffing/touching the object with the nose. Exploration time did not include the periods in which the mice were pointing their nose away from the object, even if they were beside the object, running around it, or climbing on it. To check for reliability, each observer re-scored a subset of videos in a blind fashion for each task and these scores were found to be consistent (within 10%). To determine the relative exploration of novel and familiar objects, observation scores were converted into discrimination indices (DIs) according to the formula:
To control for differences in locomotion and to assess anxious behavior, the Open Field Test was also performed in the same arena used for the behavioral testing. We used the open-source toolbox developed by Patel et al. (43) to automatically compute the total ambulatory distance as well as the amount of time spent in outer zones versus inner zones (40 × 40).
Analysis of T1AM and TA1
T1AM and its metabolites (namely TA1, thyronamine and thyroacetic acid) were assayed as previously described (27,44), with minor changes. The instrument layout consisted of an Agilent (Santa Clara, CA) 1290 UHPLC system, including a binary pump, a column oven, and a thermostatic autosampler, coupled to an AB-Sciex (Concord, Ontario, Canada) API 4000 triple quadrupole mass spectrometer, equipped with Turbo-V IonSpray source. High-performance liquid chromatography (HPLC) separation was carried out by a 2 × 50 mm, 3 μm particle size, Gemini C18 column (Phenomenex, Torrance, CA), protected by a Phenomenex Security Guard Cartridge Gemini C18 and maintained at 20°C in the column oven. The mobile phase included methanol/acetonitrile (1:4 by volume) containing 0.1% formic acid (solvent A) and water containing 0.1% formic acid (solvent B). The flow rate was 300 μL/min, and gradient conditions were as follows: 3 minutes 5% A (equilibration time), 4.5 minutes from 5% to 90% A, 0.5 minutes 90% A, and 2.5 minutes from 90% to 100% A.
Mass spectrometry acquisitions were carried out by a selected reaction monitoring-based method operating in either positive ion mode or negative ion mode, for T1AM and TA1, respectively. Three transitions were monitored for each compound (m/z: T1AM 356.2 → 195.2, 356.2 → 212.2, 356.2 → 339.1; T1AM-D4 360.2 → 199.2, 360.2 → 216.2, 360.2 → 343.1; TA1 369.1 → 127.2, 369.1 → 197.1, 369.1 → 325.3; TA1-D4 373.1 → 127.2, 373.1 → 200.1, 373.1 → 329.3), making use of optimized declustering potentials, collision energies, and collision exit potentials. Further operative parameters were set as follows: IonSpray Voltage, 5.0 and −4.2 kV; Gas Source 1, 70; Gas Source 2, 55; turbo temperature, 650°C; entrance potential, 10 V; collisionally activated dissociation (CAD) gas, nitrogen; operative pressure with CAD gas on 3.9 mPa. Calibration curves ranged from 0.25 to 100 ng/mL for T1AM and from 0.50 to 200 ng/mL for TA1.
When ACSF was assayed, each sample (0.1 mL) was spiked with 10 μL of a suitable mixture of internal standards (16 pmol of T1AM-D4 and 21 pmol of TA1-D4). Methanol (0.4 mL) was then added, and samples were shaken for 10 minutes. After centrifugation at 22,780 g for 10 min, the supernatant was dried, reconstituted with water/methanol (70/30 by volume), and injected into the HPLC-tandem mass spectrometry (HPLC-MS/MS) system. EC slices were homogenized and extracted as described (45).
TAAR1 analysis in EC slices
EC slices were fixed in 4% paraformaldehyde for three hours immediately after the experiment, then cryoprotected in 30% sucrose phosphate-buffered saline (PBS) solution, and finally cut at 25-μm thickness by using a cryostat. Slices were washed three times in PBS; treated with a solution containing 0.5% Triton X-100, 5% bovine serum albumin (BSA), and 10% donkey serum; and finally incubated with mouse anti NeuN antibody (1:50; Millipore) overnight at 4°C. The sections were subsequently washed in 0.3% Triton X-100 in PBS and incubated with a rabbit anti-TAAR1 polyclonal antibody (dilution 1:500; Abnova) for three hours at room temperature. After washing, secondary antibodies were added, namely anti-rabbit IgG Rhodamine Red X conjugated (Jakson Immunoresearch) and anti-mouse IgG conjugated with Alexa Fluor 488 (Molecular Probes) at 1:1000 dilution in 1% BSA-PBS for three hours at room temperature, washed 3 × 10 minutes in PBS, and cover slipped with Vectashield (Vector Laboratories). Serial optical sections were acquired by using an Axio Imager Z2 microscope (Carl Zeiss), and multi-channel images (transmitted fluorescence) were produced with ApoTome 2. High-resolution images were obtained by using EC Plan-NEOFLUAR 20 × /0.5 and 40 × /1.25 objectives. Analysis of TAAR1 immunofluorescence signal was performed offline on TIFF images by using MetaMorphR 5.0 r1 (Universal Imaging, Inc., Downingtown, PA). The average fluorescent staining was calculated for each image by using the quantification tool based on the detection of the contrast threshold. Three measures were obtained for each image and expressed as percentage of the area over the threshold.
For Western blot, EC slices were homogenized in a lysis buffer (10 mM Tris, pH 7.6, 100 mM NaCl, 1% Triton X-100, 0.1% SDS) containing protease and phosphatase inhibitors (2 mM Na3VO4, 1 mM NaF, 20 mM Na4P2O7) and centrifuged at 13,000 rpm (Mikro 120 microcentrifuge, Hettich) for 10 minutes at 4°C. An equal amount of proteins (50 μg) was resolved electrophoretically on a 12% Bis-Tris gel (Bio-Rad Criterion XT). Proteins were transferred onto a polyvinylidene difluoride membrane (0.45 μm), and they were blocked for one hour at room temperature in Odyssey blocking buffer (LI-COR Biosciences). To evaluate the presence of TAAR1, membranes were probed with primary antibodies against TAAR1 (1: dilution 1:1000; Abcam), at 4°C overnight in a solution of Odyssey blocking buffer and PBS with 0.2% Tween-20 (1:1). After washing, the membranes were incubated with anti-rabbit IRDye 800CW secondary antibody (926-32211; LI-COR Biosciences) for one hour at room temperature in a solution of Odyssey blocking buffer and PBS with 0.2% Tween-20 (1:1). β-Tubulin (#2128; CellSignaling) was used as a house-keeping protein. Blots were imaged with an LI-COR infrared imaging system (OdysseyCLx; LI-COR Biosciences), and densitometry analyses were performed by using Image Studio Lite v4.0 software (LI-COR Biosciences).
Statistical analysis
Data are reported as mean ± standard error of the mean. In electrophysiological experiments, comparisons between experimental groups or between FP amplitudes at different time points were performed by two-way repeated-measures ANOVA with pair wise multiple comparison procedures (Holm–Sidak method, Sigmaplot 12.0). In the other experiments, one-way ANOVA was applied. One-sample t tests was used to determine whether the DI was different from zero in the behavioral tests. Differences were considered as significant when p < 0.05.
Results
TAAR1 and T1AM are present in the EC
Western blot analysis showed TAAR1 expression in the EC, in both WT and mhAPP mice, with no significant difference in signal intensity between the two groups (Fig. 1A, B). Co-localization of TAAR1 with the neuronal marker NeuN was observed by double immunostaining, in both mhAPP and WT EC slices (Fig. 1C). Quantification of immunofluorescent staining did not reveal any significant difference between WT and mhAPP mice (Fig. 1D). Representative HPLC-MS/MS chromatograms of EC homogenate are shown in Figure 2A. A clear T1AM signal was apparent, as shown by the presence of all transitions with appropriate ratios. The endogenous T1AM concentrations we measured in our sample were in the order of a few pmoles per g of wet tissue, and the difference between WT and mhAPP mice did not reach statistical significance (5.71 ± 0.78 vs. 4.39 ± 0.1 pmol/g, p = 0.072; Fig. 2B).

TAAR1 expression in EC. (

Endogenous T1AM in EC. (
T1AM acute perfusion counteracts Aβ-induced inhibition of LTP
We focused our studies on the EC, one of the earliest affected brain regions in AD and a cortical area whose synaptic activity is negatively affected by Aβ(1 –42). As reported in Figure 3A, we observed that T1AM, at a concentration up to 5 μM, did not alter the input–output curve with respect to vehicle-treated slices. The same concentration of T1AM was applied to EC slices for 10 minutes, starting 5 minutes before HFS delivery, and we did not observe any significant change in the magnitude of LTP (130% ± 8% vs. 132% ± 6% of baseline; Fig. 3B). Then, we tested whether 5 μM T1AM could rescue LTP in EC slices treated with 200 nM oligomeric Aβ(1 –42). Indeed, this was the case, as a statistically significant difference between groups (p = 0.004) was observed (Fig. 3C). In line with previous reports (33), Aβ(1 –42) administration inhibited LTP, whereas the co-administration of T1AM restored it. In fact, the mean LTP was significantly higher in slices perfused with T1AM and Aβ(1 –42) with respect to slices treated with Aβ(1 –42) alone (127% ± 8% vs. 104% ± 2% of baseline, p = 0.001) and was comparable to that observed in vehicle-treated slices. As a control experiment, we also investigated the effect of T3, the classical TH and putative T1AM precursor. Acute perfusion with T3 (5 μM) had no effect on basic synaptic transmission and LTP expression (Fig. 3A, B), and, when T3 was co-administered with Aβ, it did not revert Aβ-induced synaptic plasticity impairment (Fig. 3C).
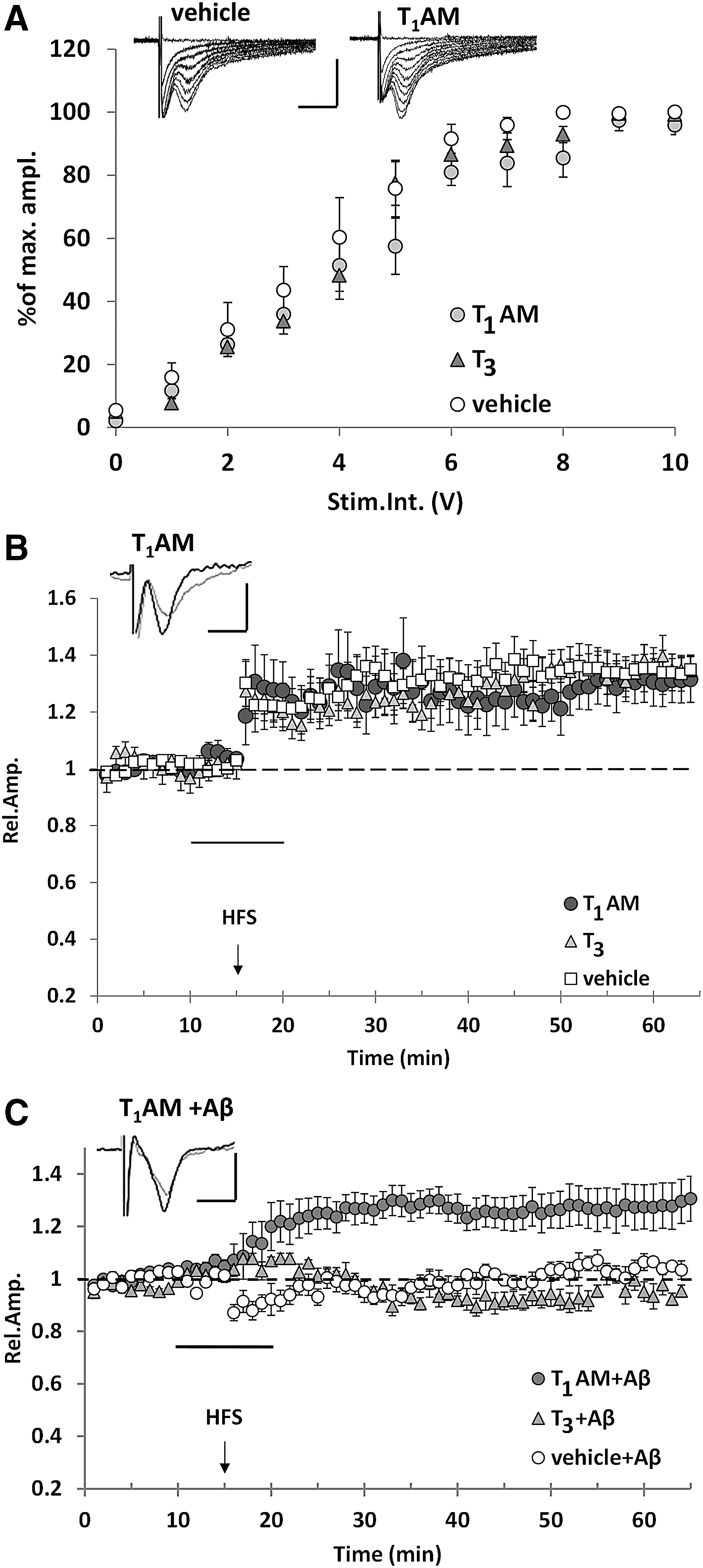
T1AM rescues the inhibitory effect of Aβ on LTP in EC slices. (
Due to the complexity of protein binding, cellular uptake, and tissue metabolism (46), nominal T1AM concentration does not necessarily reflect the concentration achieved at the receptor level. To get a better estimate of the latter, in parallel experiments, we assayed T1AM and its metabolites (namely: TA1, thyronamine, and thyroacetic acid) in the perfusion buffer eluted from EC slices, which is usually assumed to be in equilibrium with the extracellular fluid. As shown in Figure 4A, during T1AM infusion its concentration (averaged over 5–15 minutes periods) ranged from 107 ± 2 to 298 ± 39 nM, and decreased in the washout phase, reaching 44 ± 10 nM in the 40–55 minutes interval. Among T1AM metabolites, only TA1 was detected. Its concentration peaked at 198 ± 51 nM and decreased to 137 ± 22 nM at the end of the washout phase. Thus, exogenous T1AM was taken up and transformed into its main metabolite during the experiment.

TA1 administration does not rescue LTP in Aβ-treated slices. (
Since TA1 has been suggested to mediate some neurological effects induced by T1AM administration (28), and to have neuroprotective properties in a model of kainate toxicity (47), we tested the effect of 5 μM TA1 on Aβ-induced inhibition of LTP in the EC (Fig. 4B). Similar to T1AM, TA1 did not affect LTP in EC slices. However, in contrast to T1AM, we observed that TA1 administration did not rescue LTP in EC slices after exposure to Aβ (mean FPs amplitude was 96% ± 6% of baseline vs. 122% ± 6% in slices treated with TA1 alone, p = 0.035).
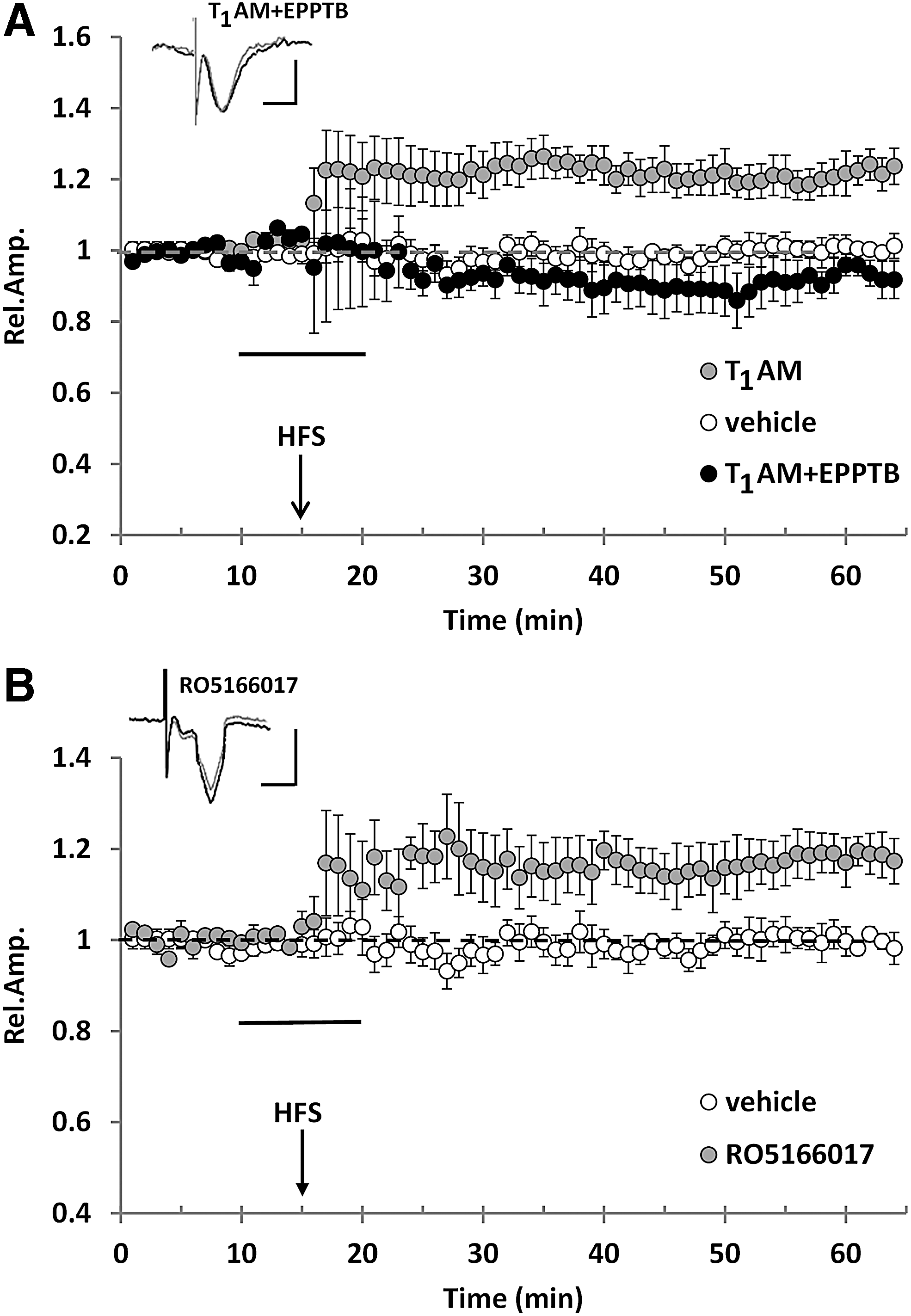
TAAR1 contributes to the protective effects of T1AM against Aβ-induced toxicity
T1AM is known to be a high-affinity agonist of TAAR1 (EC50 = 112 nM in the mouse); however, it also has additional molecular targets, including other aminergic receptors, transient receptor potential channels, and membrane transporters (46,48). To determine whether TAAR1 mediates T1AM-induced rescue of synaptic dysfunction, we used a selective antagonist (EPPTB) and a selective agonist (RO5166017) of TAAR1. EPPTB was used at a concentration that was demonstrated not to modify LTP magnitude in control experiments (5 nM) (Fig. 5A). When T1AM (5 μM) was co-perfused with EPPTB in the presence of Aβ, LTP was not rescued (99% ± 6% vs. 102% ± 3% with vehicle + Aβ and 129% ± 8% with T1AM + Aβ, p = 0.001), although EPPTB alone did not modify the response to Aβ (Fig. 5B). Then, we assessed the effects of RO5166017, a synthetic high-affinity high-selectivity TAAR1 agonist (24). The perfusion of WT EC slices with 250 nM RO5166017 did not induce a significant change in LTP expression (Fig. 5A), but the application of 250 nM RO5166017 restored LTP in Aβ-treated slices; indeed, LTP magnitude in RO5166017-treated EC slices was significantly different from that recorded in slices perfused with Aβ alone (141% ± 8% vs. 105% ± 2%, p = 0.002). As observed in the case of T1AM, the response to RO5166017 was prevented by EPPTB co-perfusion (Fig. 5C). We also had the opportunity to access a few TAAR1 KO mice, and we performed ex vivo electrophysiological recordings from the same circuitry evaluated in the previous experiments. As shown in Figure 6, LTP was significantly impaired in EC slices taken from TAAR1 KO mice compared with control mice (n = 3). These results are consistent with the hypothesis that TAAR1 mediates T1AM effects on Aβ-induced impairment of LTP, and that the T1AM-TAAR1 system may represent a new signaling pathway that rescues LTP after exposure to Aβ.

TAAR1 mediates the protective effect of T1AM against Aβ-induced inhibition of LTP in EC slices. (
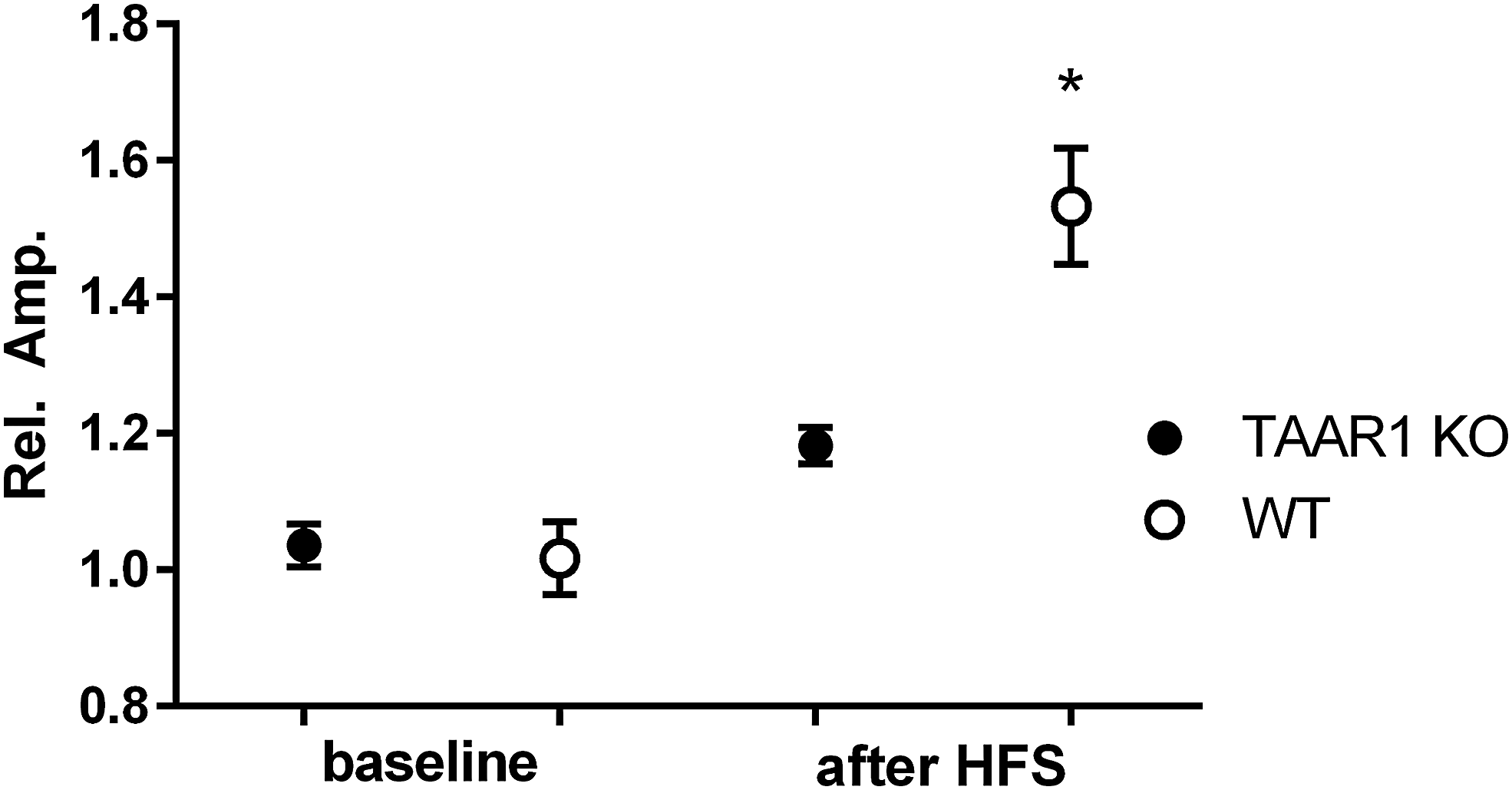
Results of ex vivo electrophysiological experiments performed to assess LTP in EC slices derived from five heterozygous TAAR1 KO mice (TAAR1 KO, number of EC slices = 6), and three WT littermates (number of EC slices = 3). Data represent mean ± SEM of the amplitude after HFS normalized to baseline amplitude and averaged over 40 minutes after HFS. *p < 0.05 versus TAAR1 KO after HFS, by two-way ANOVA and Tukey's test. KO, knockout.
T1AM counteracts early synaptic plasticity impairment in mhAPP mice
The results presented earlier prompted us to extend the investigation on the protective role of T1AM and TAAR1 to a mouse model characterized by progressive accumulation of Aβ. Specifically, we used the AD transgenic mouse model (mhAPP, J20-line), overexpressing human APP-bearing mutations linked to familial AD (49). We tested the hypothesis that T1AM could rescue LTP expression, which is impaired in this model in EC beginning at two months of age (35).
FPs recordings confirmed that HFS of the EC layer II did not induce LTP in slices taken from two-month-old mhAPP mice (mean amplitude 100% ± 4% of baseline; Fig. 7A) and that T1AM (5 μM) perfusion rescued LTP (120% ± 4% of baseline, p = 0.001 vs. vehicle-treated mhAPP; Fig. 7A). Further, T1AM effect was inhibited in co-perfusion with TAAR1 antagonist EPPTB (5 nM) (90% ± 5% of baseline), whereas a significant rescue of LTP was also achieved by perfusion with the TAAR1 agonist RO5166017 (118% ± 4% of baseline, p = 0.026; Fig. 7B). These findings are in line with the results observed in WT slices exposed to exogenous Aβ, and they suggest that in the presence of APP overexpression, acute T1AM application rescues synaptic plasticity through TAAR1 activation.
T1AM-mediated activation of TAAR1 rescues LTP impairment in mhAPP EC slices at an early stage of neurodegeneration. (
Intracerebroventricular injection of T1AM ameliorates early EC-dependent behavioral impairment in mhAPP mice
Our electrophysiological findings encouraged us to explore the neuroprotective effect of T1AM in vivo. Our aim was to investigate whether the acute i.c.v. administration of T1AM in mhAPP mice could restore the ability to perform EC-dependent behavioral tasks. As a matter of fact, the lateral EC is required for associative memory tasks based on the combined elaboration of both spatial and nonspatial information (i.e., referred to contexts and object position). Spatial associative memory can be assessed with different behavioral tests that include OPRT and OPCRT, which are selectively affected by synaptic deficits in the lateral EC, for example, those produced by progressive accumulation of Aβ in mhAPP mice (35,37).
As shown in Figure 8A, no difference was found between experimental groups in terms of locomotor activity and exploration; indeed, the total distance covered during the exploration of the arena in the open field test and the time spent in exploring the center of the arena during the trials (Fig. 8B) were comparable in the different experimental groups. In the OPRT and OPCRT tasks, a statistically significant difference between groups was observed (OPRT, p = 0.003; OPCRT, p = 0.001). As reported in Figure 8C and D, the performance of vehicle-injected mhAPP mice revealed an impairment in the ability to discriminate the novel object in relation to both its position and the surrounding context. The average DIs for mhAPP mice were not significantly different with respect to what can be expected by chance for both OPRT and OPCRT (0.02 ± 0.02 and 0.00 ± 0.03; Fig. 8C, D) and they were significantly different from DIs of age-matched WT mice (p < 0.01 for OPRT and OPCRT, respectively).
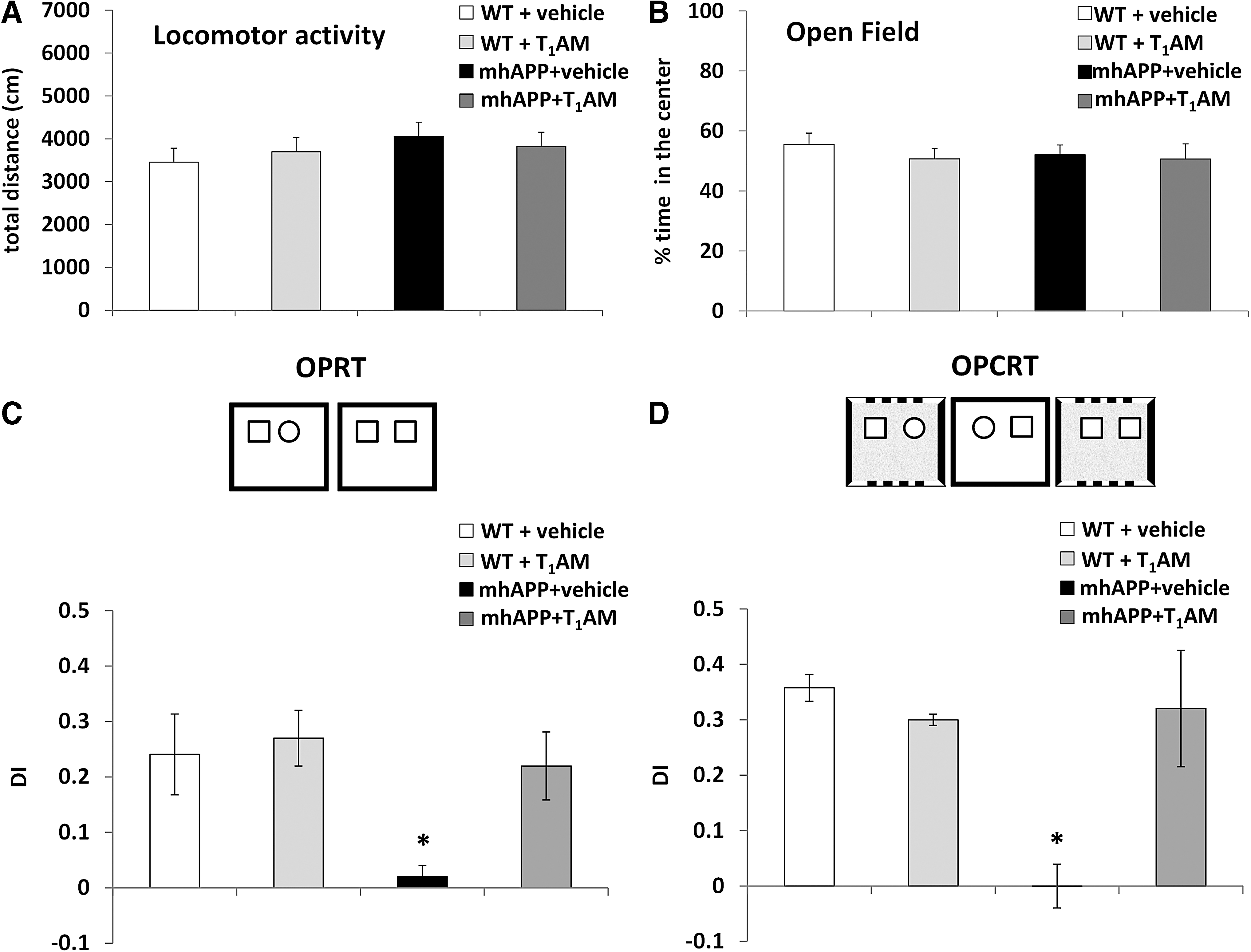
Behavioral analysis. (
In line with our electrophysiological findings, i.c.v. treatment with T1AM (0.89 μg/kg) ameliorated behavioral impairment in mhAPP mice, as mice showed a preference toward novelty not only in OPRT but also in the more complex OPCRT version of the task (Fig. 8C, D). The DIs in OPRT and OPCRT calculated for mhAPP mice treated with T1AM were significantly greater than expected by chance (0.21 ± 0.05 and 0.32 ± 0.08, p = 0.004 and p = 0.002 respectively) and significantly different from those obtained for vehicle-treated mhAPP mice (p = 0.02 and p < 0.01 for OPRT and OPCRT respectively), whereas they were comparable to those calculated for WT mice (averaging 0.24 ± 0.05 and 0.35 ± 0.02, respectively). As a control experiment, a group of WT mice was injected with T1AM and no significant difference was found between this group and WT vehicle-injected mice in both the OPRT and OPCRT (0.27 ± 0.05 and 0.30 ± 0.01). Altogether, these results indicate that T1AM i.c.v. administration ameliorates behavioral performance in mhAPP mice without any effect on locomotor activity and exploration.
Discussion
A novel insight into the complexity of TH signaling was provided by the discovery that TH derivatives represent additional chemical messengers. In particular, T1AM has been detected in rodent brain (21,44) and its administration in mice produced relevant neurological effects that are partly synergic to those induced by TH (50). Further, T1AM has been proposed as a memory enhancer as it induced pro-learning and anti-amnestic effects in mice (27,28).
We investigated the effects of T1AM on the early signs of neurodegeneration in models of Aβ toxicity, focusing our study on EC layer II. This EC layer is one of the earliest affected brain regions in AD pathology (31) and it also represents the origin of the perforant pathway, a connection that shows significant synapse loss in the early phases of AD (51,52). Further, previous studies have demonstrated a particular vulnerability of the EC to the effects of exogenously applied oligomeric Aβ(1 –42) (33,34), and early EC synaptic dysfunction has been described in a mouse model characterized by progressive accumulation of human Aβ (35).
In this study, we show that T1AM and its receptor TAAR1 are present in the EC of WT and mhAPP mice. We also demonstrate that T1AM counteracts Aβ-induced inhibition of LTP at the level of EC layer II, both when Aβ is acutely administered (Aβ 1–42 oligomers) and when it accumulates endogenously (mhAPP mice). Regarding the possible mechanism of action, T1AM interacts with different cellular targets, including TAAR1, other TAAR subtypes, monoamine transporters, adrenergic receptors, and transient receptor potential channels (53 –55). T1AM shows the highest affinity for TAAR1 (EC50 = 112 nM in mice) (21), which has been implicated in several neuropsychiatric disorders and whose activation induced pro-cognitive effects in rodent and primate models (24,56). In the present investigation, the response to RO5166017 (a selective TAAR1 agonist) and EPPTB (a selective TAAR1 antagonist) strongly suggests that the effects produced by T1AM on LTP are mediated by TAAR1, although we cannot formally exclude the possible involvement of other receptors.
To get further insight into the response to T1AM, we estimated the local concentrations of this messenger after exogenous administration. In particular, we assayed T1AM and its main metabolites in the ACSF eluted from EC slices, which is assumed to be in equilibrium with the extracellular space. During the electrophysiological recording, T1AM concentration was in the range of 40–300 nM, that is, in the same order of magnitude of the functional EC50 measured when vertebrate TAAR1 was expressed in heterologous cells (57). This is about one order of magnitude higher than the endogenous concentration detected in crude brain homogenate (27 –29,44). However, the technical problems associated with T1AM assay in biological matrices (58), and the lack of knowledge about cellular and subcellular T1AM distribution, make it difficult to compare these results. Further experiments will be necessary to determine the potential physiological or pathophysiological role of T1AM/TAAR1 signaling in the brain.
Interestingly, very low (picomolar) concentrations of Aβ have been reported to favor LTP in the hippocampus (59), although higher concentrations are detrimental for synaptic plasticity, as confirmed by our investigation in the EC. Therefore, it is not excluded that the T1AM/TAAR1 system may play a modulatory role in the response of LTP to the availability of Aβ, either under physiological conditions or in disease.
An initial approach to the evaluation of this hypothesis may be represented by the analysis of TAAR1 KO mice. Although they appear grossly normal, a neurological phenotype is actually present in homozygotes [reviewed in Rutigliano et al. (23)]. Remarkable findings have been obtained through behavioral and electrophysiological observations, which suggest increased dopaminergic drive, pointing to a putative cross-talk between TAAR1 and the dopaminergic system. However, subtle evidence of cognitive dysfunction has also been reported, and transgenic mice appeared to be slower in learning how to perform cognitive tests (56). Consistent with a potential physiological role of TAAR1 in memory and cognition, in a limited number of experiments performed on EC slices obtained from TAAR1 KO mice, we observed a significant impairment of LTP versus WT littermates. However, it is important to point out that these findings are preliminary and further evaluation will be needed to confirm the involvement of TAAR1 in LTP.
It is interesting to observe that our experiments show a trend toward a reduction in endogenous T1AM concentration and in TAAR1 expression in mhAPP mice (Figs. 1B and 2B). The possibility of a downregulation of the endogenous T1AM/TAAR1 system in this experimental model is intriguing, and it might open new pathophysiological hypotheses. However, the number of mhAPP samples available was limited and statistical significance was not achieved; therefore, further experimental work is needed to determine whether changes in this system really occur in this transgenic model.
Notably, T1AM levels in the eluate were about 16–125-fold lower than the administered dose, suggesting significant tissue metabolism and/or uptake. TA1 was the only metabolite that we could detect. Indeed, the kinetic of TA1 release was consistent with the timing of T1AM administration. Noteworthy, evidence was reported suggesting that some neurological effects elicited after T1AM administration may be actually produced by its metabolite TA1 (28,29). In the present experimental model, TA1 administration was ineffective in restoring LTP. However, it is known that T1AM can cross the plasma membrane, and deamination to TA1 may occur intracellularly (60). Therefore, we cannot exclude that some intracellular effects of T1AM-derived TA1 may not be reproduced by the administration of exogenous TA1.
We also aimed at evaluating whether T3, the putative precursor of T1AM, could reproduce T1AM effects. Although T1AM is supposed to derive from T3, the use of the latter was ineffective in preventing Aβ-induced inhibition of LTP and this may be due to the low rate of local T3 to T1AM conversion.
Our in vitro results encouraged us to investigate the effects of T1AM treatment in vivo. In a previous investigation, we showed that in mhAPP mice, early EC synaptic dysfunction is associated with behavioral deficits in associative memory tasks that require intact EC function (35,36). T1AM i.c.v. administration was performed at a dosage previously shown to induce behavioral effects and to increase tissue T1AM levels by about one order of magnitude over the baseline (27,61). The treatment was able to ameliorate associative memory in mhAPP mice at an early stage of neurodegeneration, as assessed through the OPRT and OPCRT tests. This finding is consistent with the results of the electrophysiological studies, further supports the protective role of T1AM in AD models, and may potentially open new therapeutic perspectives based on the T1AM-TAAR1 pathway. Interestingly, there is evidence that the cognitive impairment produced by scopolamine can also be rescued by T1AM (27,62).
We limited our study to the identification of the main cell surface target mediating the neuronal effects of T1AM in Aβ-induced neuronal impairment and did not evaluate the intracellular pathways responsible for T1AM effects. However, activation of stress-related protein kinases, such as JNK and p38 MAPK, appears to be a key event in Aβ-dependent neuronal impairment. In particular, these kinases are strongly activated in EC slices exposed to high levels of Aβ (33,34,63) and their level of phosphorylation is increased in the EC of mhAPP mice (35). It is noteworthy that both JNK and p38 MAPK inhibition prevented Aβ-induced synaptic plasticity impairment in hippocampal (64) and EC slices (33,34), and they ameliorated behavioral deficits in mhAPP mice (35,65). Conversely, the T1AM-TAAR1 axis can activate intracellular pathways, ultimately leading to the increase in ERK1/2 phosphorylation and c-fos expression (27,66,67) that have been demonstrated to play a fundamental role in LTP mechanisms and in memory processes (68,69). Therefore, T1AM neuroprotection may be achieved through the modulation of intracellular pathways counteracting cell stress signaling. In any case, the quick T1AM metabolism shown in Figure 4 suggests that T1AM acts by triggering a cascade of events whose final effects persist even after its concentration is normalized.
It must be acknowledged that our investigation has several limitations and further experimental work will be necessary to unravel its implications on the elusive links existing between TH signaling, Aβ toxicity, and AD. First of all, Aβ accumulation is a pathological hallmark of AD, but its causal role in the development and progression of this disease is still controversial (70,71), and the mhAPP mouse cannot be regarded as a standard model of AD. In addition, although LTP is one of the major basic mechanisms of memory and it is affected at an early stage in models of Aβ toxicity, the link between LTP impairment and cognitive dysfunction is obscure, since the latter is likely to represent the final outcome of a complex and still largely unknown pathophysiological process.
On the other hand, the role of T1AM in the context of TH-induced neuroprotection requires further investigation. Alterations in thyroid function have been linked to the pathogenesis of AD and other dementias. Evidence coming from preclinical studies suggests that TH may modulate APP gene splicing and protein processing, inducing a reduction in the synthesis of Aβ(1 –42) oligomers (14), which represent the main soluble species that induce neuronal impairment in the early stages of AD (72). With regard to clinical studies, the assay of TH levels in serum and cerebrospinal fluid showed that both subclinical hypothyroidism and hyperthyroidism represent risk factors for AD (73 –76). T1AM allegedly derives from T3 (46), and now we report that some protective properties of T1AM are not reproduced by T3. However, at present, there is no evidence that local or systemic TH administration may increase brain T1AM level, nor that the putative beneficial effects of T3 are reproduced by T1AM.
In conclusion, our study supports the concept that T1AM and TAAR1 are part of an endogenous system that can be modulated to prevent synaptic and behavioral deficits associated with Aβ-toxicity. Since T1AM and synthetic analogues appear to elicit pro-learning effects also after systemic administration (67), our results encourage further investigations aimed at determining whether the development of TAAR1 agonists may represent a novel strategy for the treatment of Aβ-related neurodegenerative disorders (77).
Footnotes
Acknowledgments
The authors gratefully acknowledge R. Di Renzo for technical assistance, Dr. F. Biondi for the excellent animal care, Dr F. Tozzi for helping with the graphical elaboration, and Dr. S. Espinoza for providing TAAR1 knockout mice.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the CNR Research Project Nanomax-Nanobrain (N.O.), and by a grant from the University of Pisa (PRA 2018 to R.Z.).