
Abstract
Background:
Graves' disease, caused by autoantibodies that activate the thyrotropin (TSH) receptor (TSHR), has only been reported in humans. Thyroiditis-prone NOD.H2h4 mice develop autoantibodies to thyroglobulin (Tg) and thyroid peroxidase (TPO) but not to the TSHR. Evidence supports the importance of the shed TSHR A-subunit in the initiation and/or amplification of the autoimmune response to the holoreceptor. Cells expressing the gene for the isolated A-subunit secrete A-subunit protein, a surrogate for holoreceptor A-subunit shedding. NOD.H2h4 mice with the human TSHR A-subunit targeted to the thyroid (a “self” antigen in such transgenic (Tgic) animals), unlike their wild-type (wt) siblings, spontaneously develop pathogenic TSHR antibodies to the human-TSH holoreceptor. These autoantibodies do not recognize the endogenous mouse-TSH holoreceptor and do not cause hyperthyroidism.
Methods:
We have now generated NOD.H2h4 mice with the mouse-TSHR A-subunit transgene targeted to the thyroid. Tgic mice and wt littermates were compared for intrathyroidal expression of the mouse A-subunit. Sera from six-month-old mice were tested for the presence of autoantibodies to Tg and TPO as well as for pathogenic TSHR antibodies (TSH binding inhibition, bioassay for thyroid stimulating antibodies) and nonpathogenic TSHR antibodies (ELISA).
Results:
Expression of the mouse TSHR A-subunit transgene in the thyroid was confirmed by real-time polymerase chain reaction in the Tgics and had no effect on the spontaneous development of autoantibodies to Tg or TPO. However, unlike the same NOD.H2h4 strain with the human-TSHR A-subunit target to the thyroid, mice expressing intrathyroidal mouse-TSHR A subunit failed to develop either pathogenic or nonpathogenic TSHR antibodies. The mouse TSHR A-subunit differs from the human TSHR A-subunit in terms of its amino acid sequence and has one less glycosylation site than the human TSHR A-subunit.
Conclusions:
Multiple genetic and environmental factors contribute to the pathogenesis of Graves' disease. The present study suggests that the TSHR A-subunit structure (possibly including posttranslational modification such as glycosylation) may explain, in part, why Graves' disease only develops in humans.
Introduction
Hyperthyroidism in Graves' disease is caused by autoantibodies that stimulate the thyrotropin receptor (TSHR) [reviewed in Ref. (1)]. Multiple genetic and environmental factors contribute to the pathogenesis of this disease, the former [reviewed in Ref. (2)], including TSHR gene single-nucleotide polymorphisms that influence intrathymic TSHR expression, thereby altering central tolerance toward the TSHR (3,4). Although a relatively common disease in humans, affecting ∼1% (particularly women) of the population in their lifetimes (5), to date there are no reports of Graves' disease occurring in other animals. Even great apes, closest relatives of humans, do not appear to develop Graves' disease (6,7). In contrast, autoimmune thyroiditis (Hashimoto's disease in humans) develops spontaneously in diverse species, including chickens, rats, mice, dogs, and marmosets [reviewed in Ref. (8)]. Of these species, NOD.H2h4 mice are presently the most extensively studied and provide a valuable model for human disease [reviewed in Ref. (9)]. In this strain, self-tolerance is broken to thyroglobulin (Tg) and thyroid peroxidase (TPO) and autoantibodies to these two thyroid autoantigens develop in association with thyroiditis (10 –13). However, there is no evidence that NOD.H2h4 mice spontaneously develop autoantibodies to the TSHR. Recently, a transgenic (Tgic) variant of NOD.H2h4 mice (hTSHR/NOD.H2h4 ) has been created in which pathogenic, stimulatory TSHR autoantibodies arise spontaneously, the transgene expressed in the thyroid being the A-subunit of the human TSHR (14).
The foregoing findings raise the question of why NOD.H2h4 mice break tolerance to Tg, TPO, and to the human TSHR A (a “self” antigen in the Tgic) but not to the endogenous mouse TSHR. One possibility is the difference between the TSHR components as well as their levels of expression. Many TSHR on the cell surface cleave into A- and B-subunits that remain attached by disulfide bonds (15,16). Some TSHR A-subunits are shed from the cell surface of living thyroid cells by a presently uncertain mechanism (17,18). There is strong evidence that the TSHR A-subunit, not the holoreceptor, is involved in the initiation and/or amplification of the autoimmune response to the holoreceptor [(19,20), reviewed in Ref. (21)]. Cells stably expressing the gene for the isolated A-subunit do not retain this protein intracellularly, but largely secrete it into the medium (22). For this reason, intrathyroidal expression of human A-subunits by the transgene in NOD.H2h4 mice is a surrogate for A-subunit shedding by the holoreceptor. Incidentally, it has recently been suggested that a human TSHR mRNA variant could similarly modulate the immune response to the TSHR or could attenuate thyroid stimulating antibody (TSAb) activity by functioning as a ligand sink (23). However, this protein is not secreted and requires cell death for release into the circulation, possibly because it is smaller than the secreted A-subunit (amino acids 22–231 vs. 22–289; numbering of both including the deleted signal peptide).
A further difference between wild-type (wt) and human A-subunit Tgic NOD.H2h4 mice is the level of A-subunit shedding from the TSH holoreceptor versus secretion of this protein. The TSH holoreceptor is expressed at a very low level in thyrocytes in vivo, whereas the human A-subunit transgene is driven by a powerful Tg promoter (14,24). In the present study, we tested the hypothesis that insufficient A-subunit is normally available to stimulate the immune response to the endogenous mouse TSHR. For this purpose, we generated Tgic mice on the NOD.H2h4 background expressing the mouse TSHR A-subunit in the thyroid.
Materials and Methods
Generating Tgic NOD.H2h4 mice expressing the mouse TSHR A-subunit in the thyroid
The transgene was constructed as previously described (24). Briefly, mouse TSHR A-subunit cDNA (amino acid residues 1–289) with a 6-histidine tag at the C terminus (22) was subcloned into the EcoRI and XbaI sites in pcDNA3 (Invitrogen, Carlsbad, CA; Life Technologies, Carlsbad, CA). This intermediate plasmid (pcDNA3-289), containing a KpnI site 5′ to the insert, was mutagenized (QuikChange; Stratagene) to introduce a KpnI site 3′ to the bovine growth hormone poly(A) tail. The bovine Tg promoter in the plasmid TgSK- (aa 2036 to 9; GenBank accession no. M35823 provided by Dr. G. Vassart, Université Libre de Bruxelles, Brussels, Belgium) was restricted with BamHI; the released 2-Kb fragment was inserted in the correct orientation into pcDNA3-289 to generate pTg-mTSHR-289.
Oocytes were obtained from NOD.H2h4 mice (originally from The Jackson Laboratory, now maintained at Cedars-Sinai Medical Center). Tg-mTSHR-289 was excised with KpnI, purified, and microinjected into NOD.H2h4 oocytes by the Mouse Genetics Core Facility, Cedars-Sinai Medical Center. From 36 pups, three founders (two males, one female) were identified by polymerase chain reaction (PCR) from genomic tail clip DNA. Offspring heterozygous for the transgene were obtained by breeding one founder male with wt female NOD.H2h4 mice. From the age of eight weeks, Tgic offspring and wt littermates were provided with drinking water supplemented with sodium iodide (0.05% NaI). Blood was drawn after 8 weeks (mice aged 4 months) and mice were euthanized after 16 weeks on iodide (aged 6 months) to harvest blood and thyroid tissues.
Embryos from mouse TSHR/NOD.H2h4 Tgics mated to wt NOD.H2h4 mice have been cryopreserved (designated mTSHR Tgic NOD.H2h4 ) by the Mouse Genetics Core Facility, Cedars-Sinai Medical Center. All mouse studies were performed with the approval of the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center and carried out with the highest standards of care.
TSHR antibody assays
TSHR antibodies were tested using three approaches:
ELISA for mouse TSHR peptide and human TSHR A-subunit protein
Peptide “mTSHR AB” corresponding to mouse TSHR amino acids 22–56, KECASPPCECHQEDDFRVTCKELHRIPSLPPSTQT, was purchased (Peptide 2.0, Chantilly, VA). This 35 amino acid peptide contains seven amino acid differences with its human counterpart. As previously described for other TSHR peptides (25), ELISA plates were coated with mTSHR A-B peptide (10 μg/mL). Recombinant human A-subunit protein (amino acid residues 22–289) secreted by Chinese Hamster Ovary (CHO) cells with an amplified transgenome (24) was purified from culture supernatants by affinity chromatography (25) and used to coat ELISA plates (5 μg/mL).
For both assays, ELISA pates were incubated with mouse sera (diluted 1: 100) followed by washing and exposure to anti-mouse IgG conjugated to horseradish peroxidase (A3673; Sigma Chemical Co., St. Louis, MO). The signal was developed with o-phenylenediamine and H2O2 and the reaction stopped using 20% H2SO4. For TSHR A-subunit protein ELISA, the positive control was serum from BALB/c mice immunized with hTSHR A-subunit adenovirus [e.g., Ref. (29)]. Monoclonal antibody (McAb) 3BD10 cross-reacted with mTSHR AB peptide and was used as a positive control in both the peptide assay and the TSHR A-subunit protein assay (25). Normal serum from BALB/c mice was used as the negative control for both assays. Data are reported as the optical density (OD) at 490 nm.
TSH binding inhibition assay
TSH binding inhibition (TBI) levels were measured in 25 μL mouse serum using a clinical assay kit (TSH receptor antibody; Kronus, Inc., Star ID). The data are reported as the % inhibition of 125I-TSH (porcine) binding to the TSH holoreceptor (extracted from porcine thyroid tissue). Because male NOD.H2h4 mice develop high serum levels of TSH (14,27), TBI was only measured in sera from female mice.
Bioassay for TSAb
A bioassay was used to measure cAMP generation by CHO cells expressing the mouse TSHR (28).
TSAb in female sera: Test serum (25 μL) + normal human serum as carrier (75 μL) was precipitated with 300 μL 20% polyethylene glycol 4000 (PEG; Sigma-Aldrich) in water. The negative controls were sera from BALB/c mice; for the positive control, BALB/c serum was supplemented with human monoclonal TSHR M22 (26) (provided by Dr. Rees Smith, Cardiff) and precipitated in the same way. The pellets were resuspended in 240 μL Ham's F12 medium supplemented with 10 mM HEPES, pH 7.4, 1 mM isobutylmethylxanthine, and 0.3% bovine serum albumin.
Purification of serum IgG: As already mentioned, TSH is elevated in the sera of male NOD.H2h4 mice on iodide (14,27) and TSH is not removed by the PEG precipitation protocol (data not shown). To avoid obtaining spurious TSAb values due to serum TSH, IgG was purified from individual male sera as follows: 60 μL test mouse serum +60 μL normal human serum was applied to Nab™ Protein A/G Spin Columns (Pierce Biotechnology, Rockford, IL) and eluted with 400 μL 0.1 M glycine, pH 2.4, into 3 fractions, each containing 40 μL 1 M Tris pH 8.5. The fraction with the highest OD value (280 nm) was pooled with 60 μL of the fraction with the second highest OD value to provide a total volume of 500 μL (“eluted IgG”). Purified IgG was precipitated with 1.5 mL 20% PEG 4000 in water and resuspended in 240 μL Ham's F12 medium supplemented with 10 mM HEPES, pH 7.4, 1 mM isobutylmethylxanthine, and 0.3% bovine serum albumin. As for PEG-precipitated sera, the positive control was purified mouse IgG supplemented with monoclonal TSHR M22 (26).
Duplicate aliquots of IgG preparations (110 μL) were applied to monolayers of mouse TSHR-expressing CHO cells (28) in 96-well plates. After incubation (90 minutes, 37°C), the medium was aspirated, intracellular cAMP was extracted with ethanol, evaporated to dryness, and resuspended in 200 μL Dulbecco's PBS. Aliquots (12 μL) were assayed using the LANCE cAMP kit (PerkinElmer, Boston, MA). TSAb activity was expressed as a percentage of cAMP values attained with IgG from BALB/c mice prepared in the same manner as the test samples.
Autoantibodies to Tg and TPO
Thyroglobulin and thyroid peroxidase antibodies (TgAb and TPOAb) were performed as reported previously [e.g., Ref. (14)]. Tg was isolated from murine thyroid glands (13); ELISA wells (Immulon 4HBX; Thermo Scientific, Rochester NY) were coated with mouse Tg (1.5 μg/mL) and incubated with test sera (duplicate aliquots, 1:100 dilution). Antibody binding was detected with horseradish peroxidase-conjugated goat anti-mouse IgG (A3673; Sigma Chemical Co., St. Louis MO), the signal developed with o-phenylenediamine, and the reaction stopped using 20% H2SO4. The negative control was serum from eight-week-old NOD.H2h4 mice on regular water; the positive control was serum from BALB/c mice immunized with mouse Tg and complete Freund's adjuvant (29). TgAb data are presented as the OD at 490 nm.
TPOAb was measured by flow cytometry using CHO cells stably expressing mouse-TPO (13). Sera (1:50 dilution) were incubated with mouse TPO-CHO cells; binding was detected with fluorescein isothiocyanate-conjugated affinity purified goat anti-mouse IgG (M30101; Invitrogen). The positive control was a previously characterized TPOAb-positive serum from NOD.H2h4 mice on iodide for 16 weeks; the negative control was serum from 8-week-old NOD.H2h4 mice on regular water. Flow cytometry was performed (10,000 events) using an FACScan with CellQuest software (Becton Dickinson, San Jose, CA). Data are reported as the geometric mean (Geo Mean).
Serum thyroxine and thyroid histology
Thyroxine (T4) levels were measured (10 μL aliquots) by ELISA (mouse/rat T4 ELISA; Calbiotech, El Cajon, CA). T4 values were computed from kit standards and expressed as μg/dL. Thyroid glands were preserved in zinc fixative (BD Pharmingen, San Diego, CA), paraffin embedded, and serial sections stained with hematoxylin and eosin (IDEXX Bioresearch Lab Animal and Biological Materials Diagnostic Testing, Columbia, MO).
Quantitative real-time PCR for thyroidal expression of the mouse TSHR A-subunit
Thyroid tissue, obtained at euthanasia, was stored in RNAlater (Life Technologies, Carlsbad, CA) for TSHR A-subunit/NOD.H2h4 (three males, three females, and non-Tgic NOD.H2h4 mice (three males, three females). Tissues were homogenized with QIAshredder columns (QIAGEN, Valencia, CA). Total RNA was prepared using the RNeasy Plus Mini kit (QIAGEN). The mRNA samples were treated with TURBO DNase (Life Technologies) to remove genomic DNA. Reverse transcription was performed with the AffinityScript QPCR cDNA Synthesis Kit (Agilent Technologies, Cedar Creek, TX) using oligo(dT) and random primers. Real-time PCR was performed using the FastStart SYBR Green Master mix (Roche, Basel, Switzerland) with 2.5% of cDNA (20 μL final volume). Reactions were run on an iCycler Thermal Cycler with the iQ5 Real-Time PCR Detection System module (BioRad Laboratories, Hercules, CA). An initial denaturation step at 95°C (10 minutes) was followed by denaturation at 95°C (30 seconds) and annealing and extension at 55°C (30 seconds) for 40 cycles. Primers for the mTSHR A-subunit, designed to avoid overlap with the endogenous mouse TSHR, were as follows: sense 5′-TCCCTGACTTGACCAAAATTTATTCCACGG-3′, mTSHR A_subunit antisense 5′-CTCGAGTTATCAGTGATGGTGGTGGTGATG-3′ (Eurofins Genomics, Louisville, KY). The endogenous mouse TSHR, Tg, TPO, and GADPH were measured using RT2 qPCR Primer Assay for mouse Tshr, mouse Tg, mouse TPO, and mouse GADPH (Qiagen).
Relative gene expression levels were calculated using the comparative Ct method (ΔΔCt), according to the Pfäffl model (30), using Bio_Rad iQ5 2.0 software. Samples were tested in triplicate; parallel controls lacked reverse transcriptase. Data were normalized to mouse GADPH.
Statistics
Significant differences between responses in different groups were determined by the Mann–Whitney rank sum test or, when normally distributed, by Student's t-test. Multiple comparisons were made using analysis of variance. Tests were performed using SigmaStat (Jandel Scientific Software, San Rafael, CA).
Results
Transgene expression of the mouseTSHR A-subunit in NOD.H2h4 mice
Offspring heterozygous for the transgene were obtained by crossing a founder male NOD.H2h4 mouse Tgic for the mouse TSHR-A-subunit with wt NOD.H2h4 females. No offspring were obtained from two other founders. To enhance thyroid autoantibody development (as in most studies using NOD.H2h4 mice), Tgics and WT littermates were exposed from the age of eight weeks to drinking water containing iodide. Unlike immunization studies [e.g., Ref. (20)], spontaneous development of TSHR antibodies is a slow process and mice were maintained on iodide until euthanasia at six months of age.
As determined by real-time PCR, the mouse TSHR A-subunit transgene was expressed in the thyroid gland of Tgic, but not wt NOD.H2h4 mice (Fig. 1A). Thyroidal expression of the endogenous mouse TSH holoreceptor was similar in the Tgic and wt mice (Fig. 1A). Thyroidal expression of the mouse TSHR A-subunit transgene did not significantly alter the expression levels of endogenous mouse Tg and TPO (Fig. 1B). For comparison with the present study describing NOD.H2h4 Tgic for the mouse TSHR A-subunit, previously reported data (31) for NOD.H2h4 mice Tgic for the human TSHR A-subunit that spontaneously develop pathogenic TSHR autoantibodies (14) are depicted in Figure 1A. These mouse and human TSHR A-subunit expression levels are far lower than in the high expressor human TSHR A-subunit NOD.H2h4 strain that remains tolerant to this protein and does not develop pathogenic TSHR autoantibodies (31).
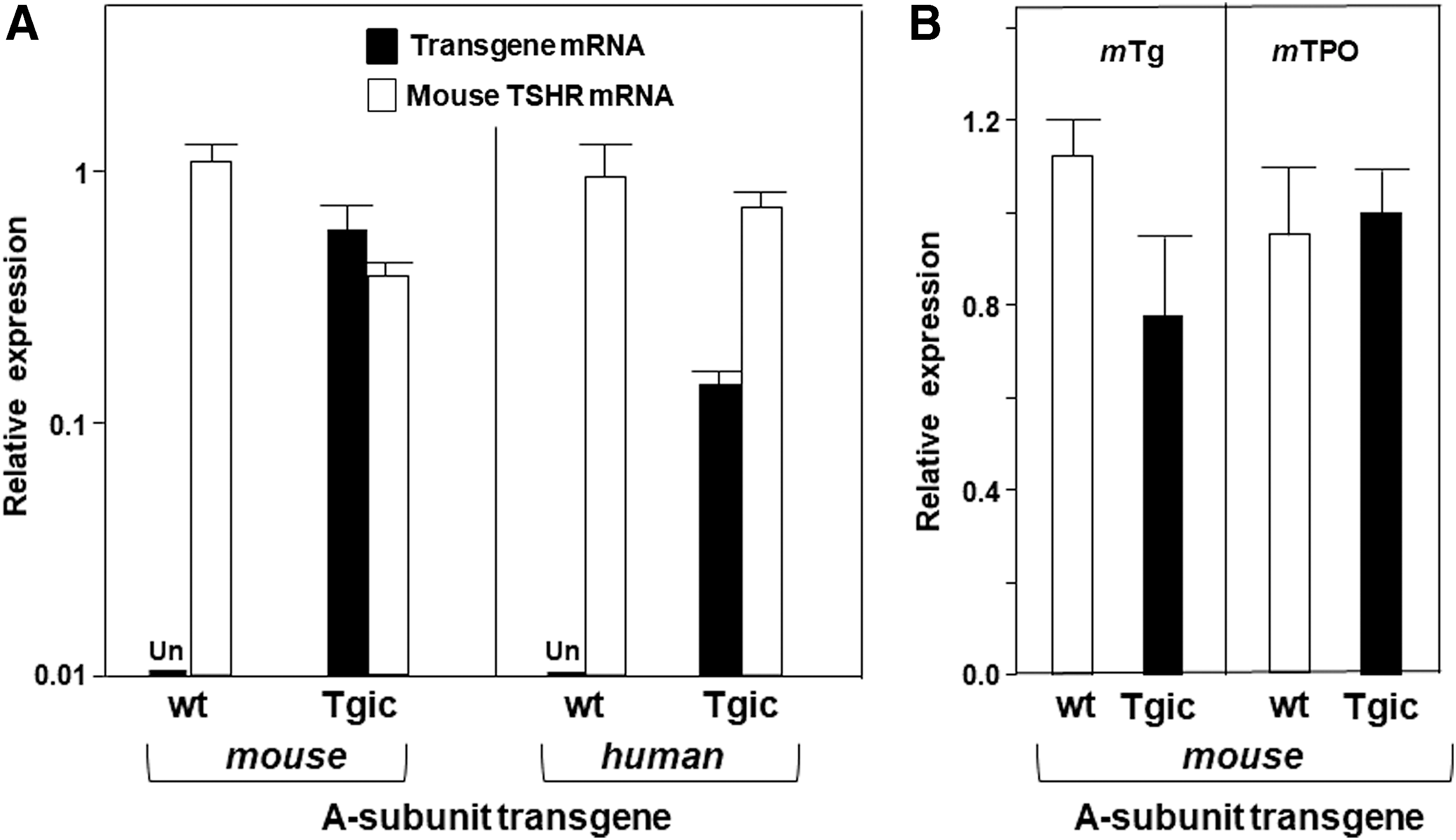
(
NOD.H2h4 mice expressing the mouse TSHR A-subunit remain euthyroid
Pathogenic TSHR autoantibodies that develop in NOD.H2h4 mice Tgic for the human TSHR A-subunit do not cause hyperthyroidism because these antibodies are to the human TSHR and do not cross react with the endogenous mouse TSHR expressed on the murine thyrocytes (14). It must be emphasized that TSHRAb arising in mice to the human TSHR, whether spontaneously or induced (e.g., using A-subunit adenovirus) activate the endogenous mouse TSHR only in some strains (such as BALB/c and C3H) but not in others (such as C57BL/6) (32). Mice of the NOD.H2h4 strain resemble C57BL/6 mice in their poor or absent recognition of the mouse TSHR by autoantibodies to the human TSHR whether induced (33) or developing spontaneously (14).
In contrast, autoantibodies arising in NOD.H2h4 mice Tgic for the mouse TSHR A-subunit (mTSHR/NOD.H2h4 ) would be expected to interact with the endogenous mouse TSHR and cause hyperthyroidism. However, this outcome did not occur. Serum T4 levels were not significantly different between Tgics and wt littermates, both male and female (Fig. 2). A few high T4 outliers were present in both Tgics and non-Tgics. The extent of thyroid lymphocytic infiltration was similar in Tgics and wt littermates with a tendency toward more infiltration in males than females (data not shown).

Serum T4 in mouse TSHR/NOD.H2h4 Tgic mice and wt littermates. Data are shown separately for individual males and females. The shading represents the mean ± 2SD for wt males or wt females separately. The numbers of mice studied are provided in parentheses. T4, thyroxine.
TSHR autoantibodies fail to develop in NOD.H2h4 mice Tgic for the mouse TSHR A-subunit
To understand the basis for the NOD.H2h4 Tgics not developing hyperthyroidism, we tested sera for TSHR autoantibodies using three different assays. It should be appreciated that unlike human (or mouse) autoantibodies to Tg or TPO, which only recognize their respective human or mouse proteins (13), human TSHRAb have long been known to cross-react with TSHR protein from other mammalian species. In particular, TSHRAb from some Graves' patients were measured as a long acting thyroid stimulator (LATS) based on their interaction with the TSHR in guinea pigs (34) or mice (35). Moreover, a clinical assay commonly used in humans to measure pathogenic TBI activity (36), which we used in our hTSHR/NOD.H2h4 mice (14), utilizes the porcine TSHR. In light of this cross-reactivity, we probed the possible development of TSHRAb in the mouse TSHR A-subunit NOD.H2h4 mice in assays using human as well as mouse TSHR.
Given the absence of hyperthyroidism, we first used ELISA because TSHR antibodies detected by this means cannot activate the TSHR and cause hyperthyroidism (37,38). Our previous ELISA studies on nonpathogenic antibodies to the human TSHR utilized recombinant human TSHR A-subunits generated in mammalian cells, as well as synthetic 20-mer peptides to the TSHR N-terminus [amino acid residues 22–41 or 37–56; termed “A” and “B,” respectively; e.g. Ref. (29)]. Because the recombinant mouse eukaryotic TSHR A-subunit protein is not available, we used a synthetic mouse TSHR peptide to the same region (termed “A–B,” residues 22–56). Sera from male and female mouse TSHR A-subunit/NOD.H2h4 Tgics aged six months (n = 21) and those of wt littermates (n = 41), were all negative (data not shown). In the off chance of species cross-reactivity, we retested the same sera with human eukaryotic TSHR A-subunits, again with negative results (data not shown).
Second, we used a clinical assay for pathogenic TSHR autoantibodies, which involves competition for TSH binding to the porcine TSH holoreceptor. For this assay, only sera from female mice were tested because of the confounding effect of high TSH levels in males (14). As with the ELISA, mouse TSHR A-subunit/NOD.H2h4 Tgics were negative (Fig. 3, left panel). For comparison, replotted data from a previously reported study on human TSHR A-subunit/NOD.H2h4 Tgics using the same assay (31) indicate positivity for some (8/18) sera in this assay (Fig. 3, right panel). These negative data for mouse TSHR A-subunit-NOD.H2h4 Tgics could be explained by lack of cross-reactivity to the porcine TSHR by antibodies to the mouse TSHR.
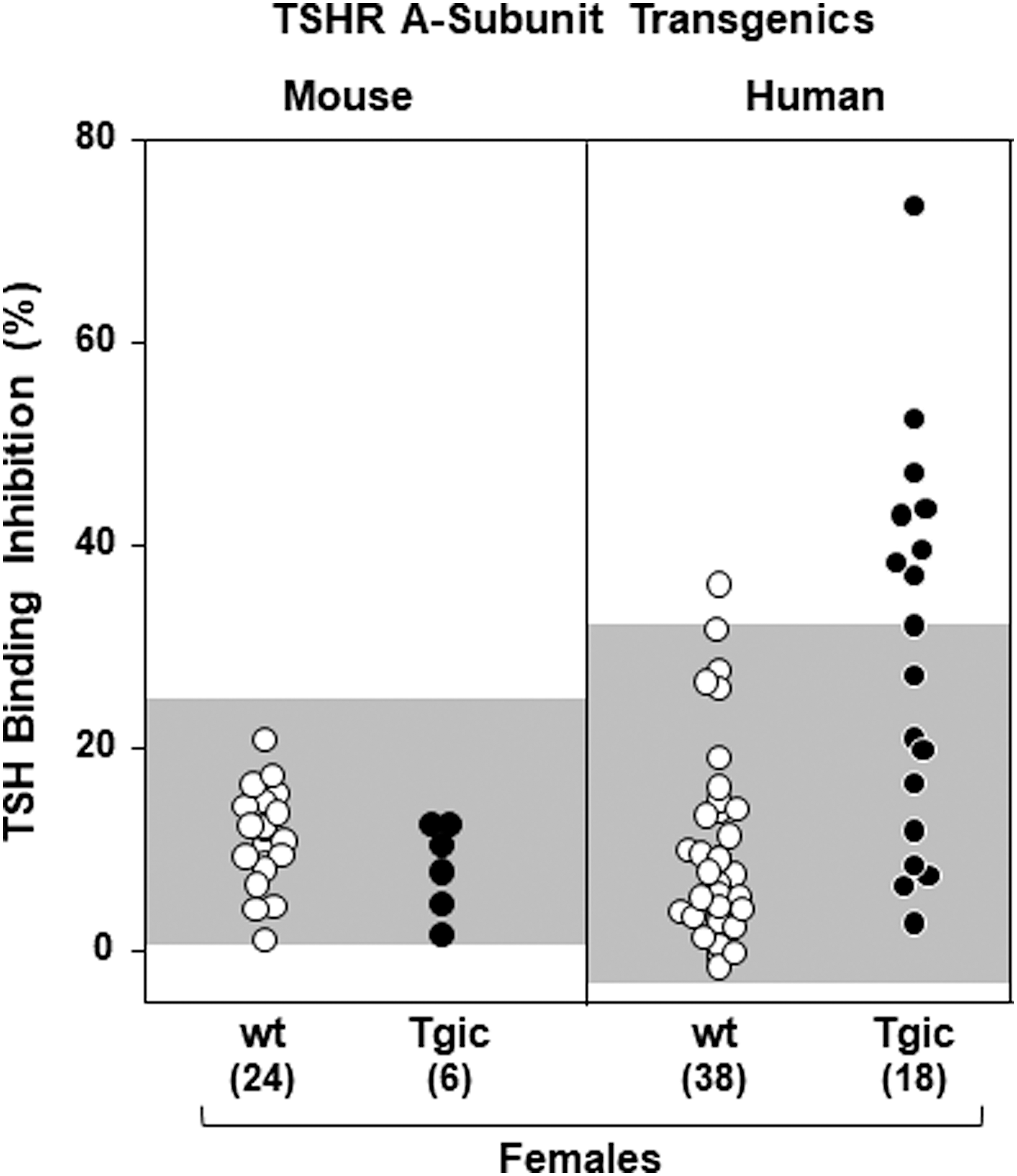
TBI activity is absent in the sera of mouse TSHR-A-subunit/NOD.H2h4 Tgic females and wt female littermates (left panel). Data are shown for individual mice. The right panel illustrates TBI activity in sera from female Tgic mice expressing the human TSHR A-subunit (hTSHR/NOD.H2h4 strain; data compiled for backcross generations N6 to N9 [from Ref. (31)]. The shading represents the mean + 2SD TBI values for wt mice. The numbers of mice studied are provided in parentheses. TBI, TSH binding inhibition.
Finally, to overcome species specificity problems of the receptor, we measured functional TSAb in a bioassay using CHO cells expressing the mouse TSHR (32). For this assay, serum IgG is precipitated using polyethylene glycol (PEG) and reconstituted in buffer. Because of the high TSH in male mice, it was necessary to first purify IgG using protein A/G before PEG precipitation. None of the sera from 12 mouse TSHR A-subunit-NOD.H2h Tgics was positive in this bioassay (Fig. 4, left panel). As expected, non-Tgic NOD.H2h4 mice yielded negative results, except for 1 spurious positive among the 32 animals studied. The strong response to human TSHR McAb M22 confirms the validity of the bioassay. For comparison with these negative responses, TSAb positivity was observed for 6/16 human TSHR A-subunit/NOD.H2h4 Tgics in an assay using CHO cells expressing the human TSHR [Fig. 4, right panel; data replotted from Ref. (14) in addition to some unpublished data described in the Figure 4 legend].
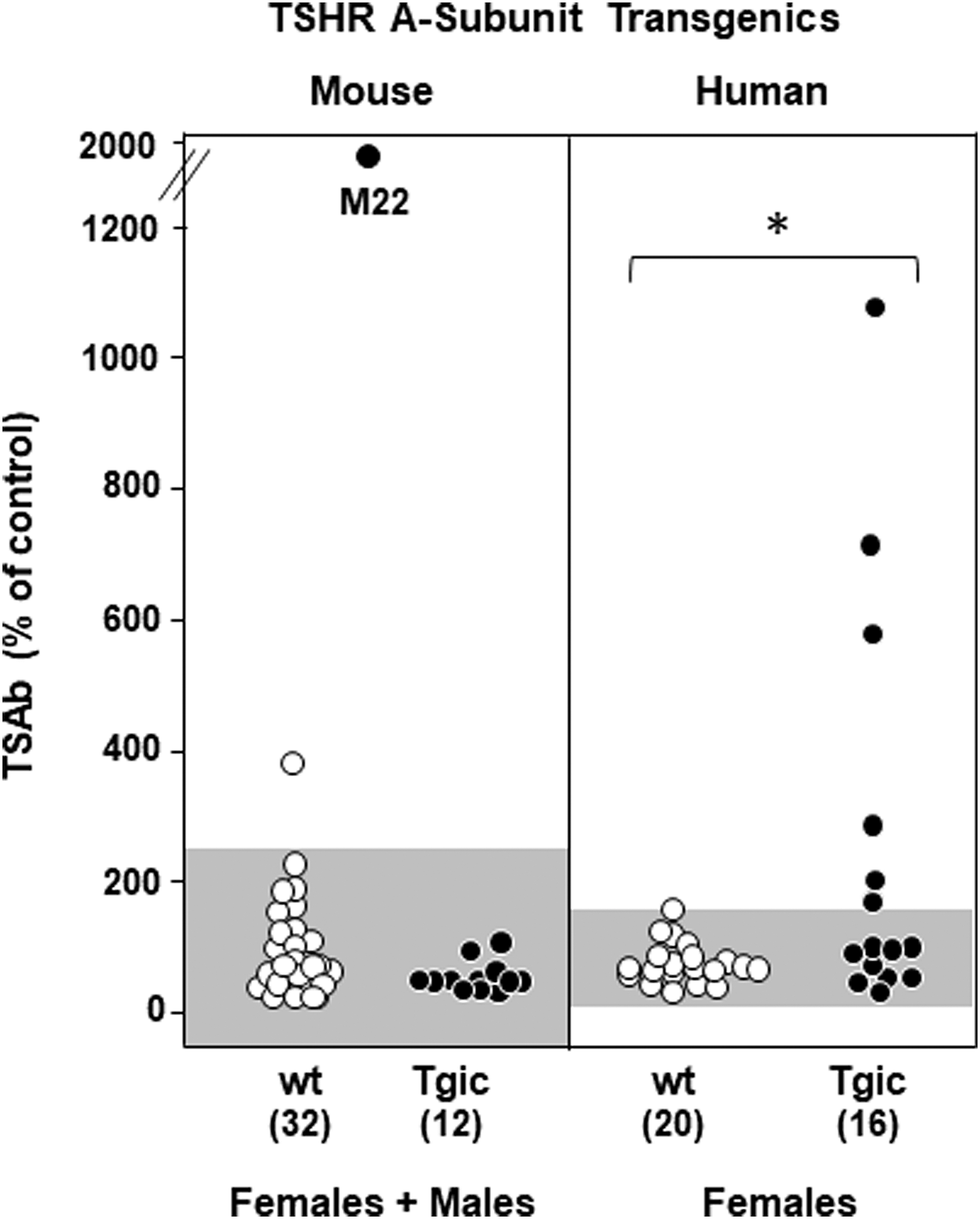
Absence of TSAb activity in mouse TSHR-A-subunit/NOD.H2h4 Tgic females or males. Data are shown for individual mice. The point indicated as “M22” is the positive control provided by a monoclonal TSAb. Included in the right panel are data for female NOD.H2h4 Tgics expressing the human TSHR A-subunit; backcross generations N6 to N8 [N6, data replotted from Ref. (14); N7 and N8 data not previously reported]. TSAb was measured in PEG-precipitated sera. For males, IgG was first purified on protein A/G columns to remove high TSH levels characteristic of this sex in wt NOD.H2h4 mice (see the Materials and Methods section). For each panel, the shading represents the mean + 2SD for wt mice. The numbers of mice studied are provided in parentheses. *p = 0.032 (Mann–Whitney rank sum test). TSAb, thyroid stimulating antibody.
It may seem paradoxical that M22 stimulates both the human and the mouse TSHR, whereas TSAb arising to the human TSHR, whether spontaneously or induced, activate the endogenous mouse TSHR only in some mouse strains (such as C3H and BALB/c) but not in others (such as C57BL/6) (32). M22 is a highly potent TSAb (26) developed by affinity maturation of human immunoglobulin germ line genes (not the same as mouse germ line genes). Moreover, M22 resembles some (not all) Graves' TSHRAb in their interaction with the mouse TSHR in the LATS assay (35). Taken together, these observations emphasize that receptor activation involves species specificity involving both the TSHR and the immunoglobulin genes from which the TSAb are derived.
Tg and TPO antibodies in mouse TSHR A-subunit Tgics
Tgic expression of the mouse TSHR A-subunit in NOD.H2h4 mice had no effect on the development of autoantibodies to Tg or TPO. Thus, as detected by ELISA using mouse Tg as antigen, TgAb levels were similar in Tgic and non-Tgic animals with no difference between the sexes (Fig. 5A). TPOAb levels determined by flow cytometry using mouse TPO-expressing CHO cells were significantly higher in female than male wt NOD.H2h4 mice (Fig. 5B, left panel), consistent with previous data (39). In Tgic NOD.H2h4 mice, the numbers were too small to achieve statistical significance between the sexes (Fig. 5B, right panel).

TgAb
Discussion
Previous studies have tested the outcome of using mouse TSHR (or the TSHR A-subunit) to immunize mice and generate TSHR antibodies specific for the endogenous mouse TSHR. Immunization with adenovirus encoding the mouse TSHR-A-subunit failed to induce TSHR antibodies in BALB/c mice, but TSHR antibodies were induced in TSHR-knockout BALB/c mice (40). In contrast, DNA vaccination with a mouse TSHR ectodomain variant induced TSHR antibodies in wt BALB/c mice (41). More recently, mouse TSHR A-subunit DNA vaccination combined with electroporation in BALB/c mice induced low levels of stimulating TSHR antibodies together with some characteristics of Graves' orbitopathy (42). Repeated DNA vaccination, particularly combined with electroporation, seems to be more effective at breaking self-tolerance to the mouse TSHR than adenovirus immunization.
Our goal was to determine whether we could develop a mouse strain that, without intervention such as microorganisms, spontaneously breaks self-tolerance and develops antibodies to the mouse TSHR. For this purpose, we generated Tgic NOD.H2h4 mice expressing the mouse TSHR A-subunit in the thyroid gland. The reason for this approach was twofold: first, the success of spontaneous TSHR antibody generation by expressing the human TSHR A-subunit in the thyroid of NOD.H2h4 Tgics expressing the human TSHR A-subunit in the thyroid (14); second, the possibility that insufficient TSHR A-subunit was shed from the endogenous mouse TSHR to initiate the immune response. The latter possibility was supported by our observation that injecting recombinant human TSHR A-subunit protein into hTSHR/NOD.H2h4 mice enhanced the development of pathogenic TSHR antibodies (38). It must be emphasized that in Tgic mice, the human- or the mouse-TSHR A-subunits are not foreign proteins. Both proteins represent self-antigens.
Of the three founder mouse TSHR-A-subunit Tgics, we obtained heterozygous mTSHR/NOD.H2h4 offspring from one male. To enhance development of thyroid autoantibodies (as in studies from others and ourselves), Tgics and wt littermates were exposed from the age of eight weeks to drinking water containing iodide. Spontaneous development of TSHR antibodies is a slow process and mice were euthanized after a four-month exposure to iodide (six months of age). This time interval was based on our previous studies using hTSHR/NOD.H2h4 Tgics showing that TSHRAb were undetectable in sera drawn from mice exposed to iodide for only two months (43,44). These studies contrast with immunization in which TSHRAb can be detected 1 week after 2 TSHR A-subunit adenovirus injections 3 weeks apart, namely in mice aged 2.5 months (33). Time limitations, together with the length of time required for the spontaneous development of TSHRAb (four months on iodide from the age of two months), restricted the number of Tgic mice we could study.
None of the mouse A-subunit/NODH2h4 transgenics developed TSHRAb when tested by ELISA (nonpathogenic antibodies), or by TBI or bioassay (pathogenic antibodies). Compared with our human TSHR-A subunit Tgics that spontaneously develop TSHR antibodies, the mouse TSHR A-subunit transgene was expressed at a similar level in the thyroid. Therefore, unlike very high expressions of the human TSHR A-subunit in the thyroid (45) and thymus of high expressor hTSHR/NOD.H2h4 mice (31), such an explanation is unlikely for the inability of mouse TSHR/NOD.H2h4 Tgics to develop TSHRAb. Although we did not test transgene expression in the thymus, thymic expression levels are generally consistent with that in the peripheral organ (46). Incidentally, based on our studies in human TSHR A-subunit Tgic mice, in our opinion it is unlikely that TSHRAb developed transiently and then disappeared in the mouse TSHR A-subunit NOD.H2h4 Tgics.
Previously, we observed that immunization of TSHR knockout mice (BALB/c background) developed higher levels of pathogenic TSHR antibodies after immunization with adenovirus expressing the human as opposed to the mouse TSHR A-subunit (40). In the light of these observations, the mouse TSHR A-subunit protein appears to be less immunogenic than the human TSHR A-subunit, perhaps because of its intrinsic amino acid sequence and/or because it contains one less glycosylation site than the human TSHR A-subunit (6,40). Indeed, the most striking difference between the amino acid sequences of TSHR A-subunits in humans versus nine other placental mammalians is that, except for the great apes (chimpanzees, gorillas, and orangutans), all other mammals, namely two species of old world monkeys, swine, dogs, cats, and mice, had only four rather than five glycosylation sites (6,40). Glycosylation of proteins is important for their uptake, processing, and presentation to T cells [reviewed in Ref. (47)]. The lower levels of glycosylation may be one factor contributing to the difficulty of breaking self-tolerance to the endogenous wt mouse TSHR A-subunit, which can be achieved (as mentioned) using the adjuvant effect of adenovirus in TSHR knockout mice (40) or DNA combined with electroporation in wt mice (42).
In conclusion, for the human TSHR A-subunit, Tgic thyroidal expression of the isolated mouse TSHR A-subunit, a surrogate for increased amounts of shed A-subunit, did not lead to the spontaneous development of TSHR antibodies in autoimmune thyroiditis-susceptible NOD.H2h4 mice. A likely explanation for this finding is a structural difference in the TSHR A-subunit between the human and the mouse TSHR A-subunit, possibly involving lesser glycosylation in mouse than in the human TSHR A-subunit. Based on amino acid sequence motifs, other nonprimates also have less glycosylated TSHR A-subunits than humans. We speculate that our present observations, if extended to other nonprimates, may explain, in part, why Graves' disease only develops in humans.
Footnotes
Acknowledgments
We thank Dr. B. Rees Smith for providing us with aliquots of human stimulating monoclonal antibody M22. The study was supported by the following grants: NIH DK 54684 (S.M.M.) and DK19289 (B.R.).
Author Disclosure Statement
No competing financial interests exist.