
Abstract
Background:
The BRAFV600E mutation is the most common driver mutation in papillary thyroid cancer (PTC) and anaplastic thyroid cancer (ATC). This mutation is considered actionable and, for BRAFV600E-mutated ATC, a BRAF inhibitor (dabrafenib) in combination with an MEK inhibitor (trametinib) is FDA approved. BRAF inhibitors have also shown efficacy in BRAFV600E-mutated PTC. However, as with all targeted therapies, resistance to these drugs eventually develops. It is essential that we understand the mechanisms of resistance to the BRAF inhibitors in thyroid cancer to develop future strategies to effectively treat these patients and improve survival.
Patients:
Herein, we describe four patients with thyroid cancer treated with selective BRAF inhibitors, who developed a RAS mutation in addition to the BRAFV600E mutation at progression.
Results:
Patients 1 and 3 acquired a KRASG12V mutation in the progressive tumor, patient 2 acquired a NRASQ61K mutation in a progressive lymph node, and patient 4 acquired NRASG13D mutation on liquid biopsy performed at the time of radiographic disease progression.
Conclusion:
Similar to the melanoma experience, the emergence of RAS mutations appears to act as a mechanism of resistance to BRAF inhibitors in thyroid cancers.
Introduction
The most frequent driver mutation detected in thyroid cancer is the BRAFV600E mutation, which occurs in more than 50% of papillary thyroid cancers (PTCs) and in ∼45% of anaplastic thyroid cancers (ATCs) (1 –6). The BRAF protein is a member of the RAF kinase family. It is usually activated by growth factors via RAS and regulates the mitogen-activated protein kinase factor (MAPK) pathway to induce cell division and/or differentiation (7 –9). Activating mutations of the BRAF gene occur mostly within the kinase region of the protein and induce its constitutive activity (10).
Therapies specifically targeting BRAFV600E have been explored in BRAF-mutated PTC and ATC (11,12). Dabrafenib is a selective BRAF inhibitor that is FDA approved for BRAF-mutated melanoma, lung cancer, and ATC (13 –15), but not for PTC. Vemurafenib is also a selective BRAF inhibitor studied in thyroid cancer but it is only approved for BRAF-mutated melanoma (11). Encorafenib is the most recent FDA-approved selective BRAF inhibitor in combination with binimetinib (MEK inhibitor) for BRAF-mutated melanoma. These drugs have not yet been studied in thyroid cancer.
FDA approval of dabrafenib in BRAF-mutated ATC was based on a phase 2 clinical trial that enrolled 23 patients globally (16). In this trial, patients were treated with a combination of dabrafenib and trametinib, a kinase inhibitor targeting the MEK protein downstream of BRAF. The response rate was 69%. The median progression-free survival (PFS) and overall survival (OS) had not been met at the time of the publication and presentation to the FDA; however, a follow-up abstract presented at ESMO 2018 (17) reported a median PFS and OS of 60 and 86 weeks, respectively.
Although BRAF inhibitors are not approved for PTC, an open-label, nonrandomized phase 2 trial at 10 academic centers worldwide demonstrated antitumor activity in patients with progressive, BRAFV600E-mutated PTC treated with vemurafenib (12). Patients were divided into two groups. Cohort 1 was comprised of patients who had not received vascular endothelial growth factor (VEGFR)-targeted kinase inhibitors (n = 26), and cohort 2 included patients who had been previously treated with a VEGFR-targeted kinase inhibitor (n = 22). The partial response rate in cohort 1 was 38.5%, median PFS was 18.2 months, and the median OS was not reached. In cohort 2, the response rate was 27.3%, and median PFS and OS were 8.9 and 14.4 months, respectively. The efficacy of dabrafenib with or without trametinib was also studied in BRAF-mutated PTC in a large multicenter study. Patients were randomized to receive either single-agent dabrafenib or dual therapy with dabrafenib plus trametinib. The investigators reported similar objective responses in both groups, 42% versus 45%. The PFS was longer in the dual therapy arm (11.4 months vs. 15.1 months) but this was not statistically significant (18). Patients on the single-agent dabrafenib were allowed to cross over to dual therapy but results of the responses in this group of patients were not reported. Finally, a clinical trial using dabrafenib plus lapatinib (HER2/HER3 inhibitor) was recently reported at ASCO 2018 (19). This study enrolled all BRAF-mutated thyroid cancers, some of which had been previously treated with BRAF inhibitors. The authors reported a 58% response rate in differentiated thyroid cancer with a PFS of 18 months. Altogether, these recent data indicate that BRAF inhibitors in combination with other targeted therapies have anti-tumor activity in advanced thyroid cancers, in particular ATC.
However, as with other tumor types, thyroid cancer patients eventually develop resistance to BRAF-directed therapies. Several mechanisms of BRAF/MEK-inhibitor resistance acquired during therapy have been well described in melanoma and colorectal cancer patients. These mechanisms involve genetic and/or epigenetic alterations that reactivate the MAPK and/or the PI3K/AKT pathways. These include alternate BRAF gene splicing events, BRAF/BRAFV600E protein amplification, mutations in the PI3K pathway (PIK3CA, PTEN, PIK3R1), mutations in the MEK1/2 gene, and upregulated expression of the MET, PDGFRβ, or IGF1R receptors (20 –27). In addition, acquired mutations in the RAS gene family, in particular NRAS (17.8%), are often detected (21,26,28).
In thyroid cancers, however, the mechanisms of resistance to kinase inhibitors are less well understood. Acquisition of a KRAS mutation has been described in RET/PTC1 cells cultured long term with the anti-VEGFR inhibitor pazopanib (29). Regarding BRAFV600E-mutated cells, resistance to BRAF inhibitors can be induced by epigenetic mechanisms such as increased HER3 receptor expression and activation through an autocrine NRG1 loop (30). Also, mutations altering cell cycle- or DNA damage recognition pathways have been described (31,32). We recently demonstrated that long-term exposure (>5 months) of BRAFV600E-mutated poorly differentiated thyroid cancer (PDTC) cells to 1 μM vemurafenib in vitro promoted the emergence of an additional KRASG12D mutation (33). The presence of both mutations significantly increased the rate of proliferation of the affected cells in comparison with cells harboring the BRAFV600E mutation only.
Confirming these data, a case report in a patient on a BRAF inhibitor for BRAFV600E-mutated PTC who developed a KRASG12V mutation at progression was recently published (34). Herein, we describe four additional patients with BRAFV600E-mutated thyroid carcinomas who received BRAF-directed therapy for their disease. These four patients developed drug resistance and were found to have acquired secondary RAS mutations in the progressive tumor, some of them different from the recently reported KRASG12D and KRASG12V. IRB approval was obtained before data collection.
Case Series
Case 1 is a 60-year-old man diagnosed with PTC who underwent a total thyroidectomy and radioactive iodine (RAI) therapy 14 years before the initiation of vemurafenib. His primary tumor details are unknown. Ten years after initial diagnosis, he underwent surgical excision of a paratracheal mass with sacrifice of the right recurrent laryngeal nerve. Surgical pathology confirmed PTC in the 4.5-cm mass with extension into surgical margins. A follow-up positron emission tomography-computed tomography (CT) showed no evidence of significant disease. However, six months later, the patient reported a palpable mass in the neck. Fine needle aspirate biopsy (FNAB) revealed bilateral recurrent PTC. The patient came to our institution for a second opinion. Head and neck CT showed extensive disease in the lower neck and paratracheal region with nonspecific bilateral pulmonary nodules. He underwent bilateral neck dissection and esophageal muscularis resection. The surgical pathology confirmed PTC in 7 out of 16 lymph nodes and the mediastinal mass showing columnar features with necrosis indicating an aggressive histopathology. The Ki-67 proliferation index was increased at ∼20%. Next-generation sequencing (NGS) analysis detecting mutations in the coding sequence of 134 genes and copy number variations in 47 genes was performed on DNA from the tumor material by the MD Anderson CLIA-certified diagnostics laboratory. The tests revealed a BRAFV600E mutation.
One month later, the patient underwent intensity-modulated radiation therapy with 60 Gy to the postoperative area. Two years later, a chest CT showed a marked increase in the size of the pulmonary nodules. Given the progression, the patient was started on vemurafenib. Four months later, imaging showed significant improvement of the bilateral pulmonary nodules. This response was maintained for more than 2 years, but at 30 months later, a chest CT showed a marked increase in the size of one lung nodule while all other nodules remained stable. The patient continued on vemurafenib. Forty-five months later, progression of the lung nodule from 1.8 to 2.9 cm was evident on a chest CT. FNAB confirmed PTC histology of the progressing lung nodule. NGS was performed on the specimen, identifying the pre-existing BRAFV600E mutation, concomitant with a new KRASG12V mutation. After 50 months of vemurafenib, the drug was stopped and the patient was started on lenvatinib but he did not respond to this treatment. The patient was subsequently started on dabrafenib plus trametinib and had a partial response, which lasted ∼14 months before progression ensued.
Case 2 is a patient who was previously reported elsewhere (35) and therefore will only be described briefly. This man presented with ATC at the age of 60 years. The patient had been treated initially with cytotoxic chemotherapy but had progression of disease and then presented at our institution with a massive tumor arising from the left thyroid lobe, invading the esophagus and encasing the carotid and vertebral arteries. The presence of a BRAFV600E mutation was confirmed on liquid biopsy (VAF 26.4%) by using Guardant360™, as tumor biopsy yielded no viable cells. Other mutations included TP53R175H (VAF 22.3%), EGFRG322S (VAF 1.6%), and BRAF amplification (Table 1). He was started on dabrafenib plus trametinib. He had a rapid clinical response with relief of dyspnea and dysphagia but after four weeks developed a new palpable lymph node in the left neck. The lymph node was biopsied; NGS sequencing was performed as described earlier and was positive for both BRAFV600E and TP53R175H mutations, as well as a new NRASQ61K mutation (Table 1). Because immunohistochemistry identified a PD-L1-positive tumor micro-environment, we added pembrolizumab to his treatment regimen. The patient responded to the triple therapy (dabrafenib, trametinib, pembrolizumab, or “DTP”) and eventually had a complete resection of his primary tumor followed by adjuvant chemoradiation. The patient remains disease free on DTP and is alive 3 years after his initial diagnosis of ATC.
Overview of Missense Mutations in BRAFV600E Positive Samples at Baseline and at Progression Under BRAF Inhibitors
Indicates that the mutations were detected on liquid biopsy (cfDNA, Guardant 360).
Case 3 is a 74-year-old woman with reportedly benign thyroid nodules known for 6 years who developed rapid onset hoarseness due to vocal cord paralysis and shortness of breath leading to emergent tracheostomy. Incisional biopsy confirmed an ATC. Staging imaging studies revealed stage IVC disease with tracheal invasion, esophageal invasion, metastases to the lung, liver, bones, and a suboccipital mass. NGS identified the following mutations in her primary untreated tumor: BRAFV600E, ATMI1986V, and MCL1 (nonsense). Guardant360 liquid biopsy testing before initiation of therapy showed the following: BRAFV600E (VAF 0.4%), TP53R273H (VAF 0.4%), EGFRR776H (VAF 0.2%), NTRK1H571Y (VAF 0.6%), and MTORV169I (VAF 0.3%) (Table 1). She was treated with one dose of bridging chemotherapy (abraxane) while waiting for results of molecular testing. Dabrafenib plus trametinib was initiated after the BRAFV600E mutation was identified. She had a dramatic response to therapy within 2 months, with a decrease in size of the primary tumor from 4.3 to 1.1 cm with an almost complete metabolic response and improvement in her distant metastatic disease. This response was maintained for 11 months after initiation of dabrafenib plus trametinib, when she developed recurrence of a suboccipital cutaneous nodule; the remainder of her disease had remained stable. This area was observed but continued to progress (Fig. 1). She then underwent total thyroidectomy, bilateral central node and left lateral neck dissection, as well as surgical excision of the suboccipital nodule. Final pathology confirmed ATC with squamoid features consistent with ATC in the suboccipital nodule. The NGS of the suboccipital nodule identified the following somatic mutations: BRAFV600E, KRASG12V, TERT 124 C>T, ATMI1986V, and AKT2E17K; no fusions were identified (Table 1). The patient declined chemoradiation, was restarted on dabrafenib and trametinib immediately after surgery, and received one dose of pembrolizumab. She was lost to follow-up but was alive with no evidence of progression 9 months postoperatively and 24 months after her initial diagnosis.

Patient 3's F18-FDG PET/CT axial image through the neck (
Case 4 is a 50-year-old man who underwent total thyroidectomy for a 4.5-cm PTC, with extrathyroidal extension into skeletal muscle. CLIA-targeted sequencing identified the BRAFV600E mutation in his primary untreated tumor. He received postoperative RAI treatment on two occasions 8 months apart with a cumulative activity of 255 mCi 131I. One year after diagnosis, he was found to have a solitary 2-cm lung metastasis; a biopsy confirmed PTC. He was followed with serial images and 1 year later he developed recurrence in the neck with development of a new 1-cm left tracheoesophageal groove mass. Although it was initially stable for 4 months, it then rapidly progressed to a 4-cm mass with involvement of the trachea and larynx. His lung metastases remained stable. FNAB of the neck mass showed recurrent PTC. He was started on dabrafenib plus trametinib, but one week later developed worsening shortness of breath and stridor requiring tracheostomy. He continued on systemic therapy with several interruptions and dose reductions due to fever, chills, and arthralgia. Restaging exams demonstrated anatomic and metabolic response to therapy. This response was sustained for 20 months, when progression in the neck was noted. Pembrolizumab was added to his regimen, but despite three infusions, his neck disease continued to progress (Fig. 2). FNAB of the progressive neck disease showed PTC. A liquid biopsy performed at the time of progression on triple therapy with dabrafenib, trametinib, and pembrolizumab identified BRAFV600E and NRASG13D mutations (Table 1).
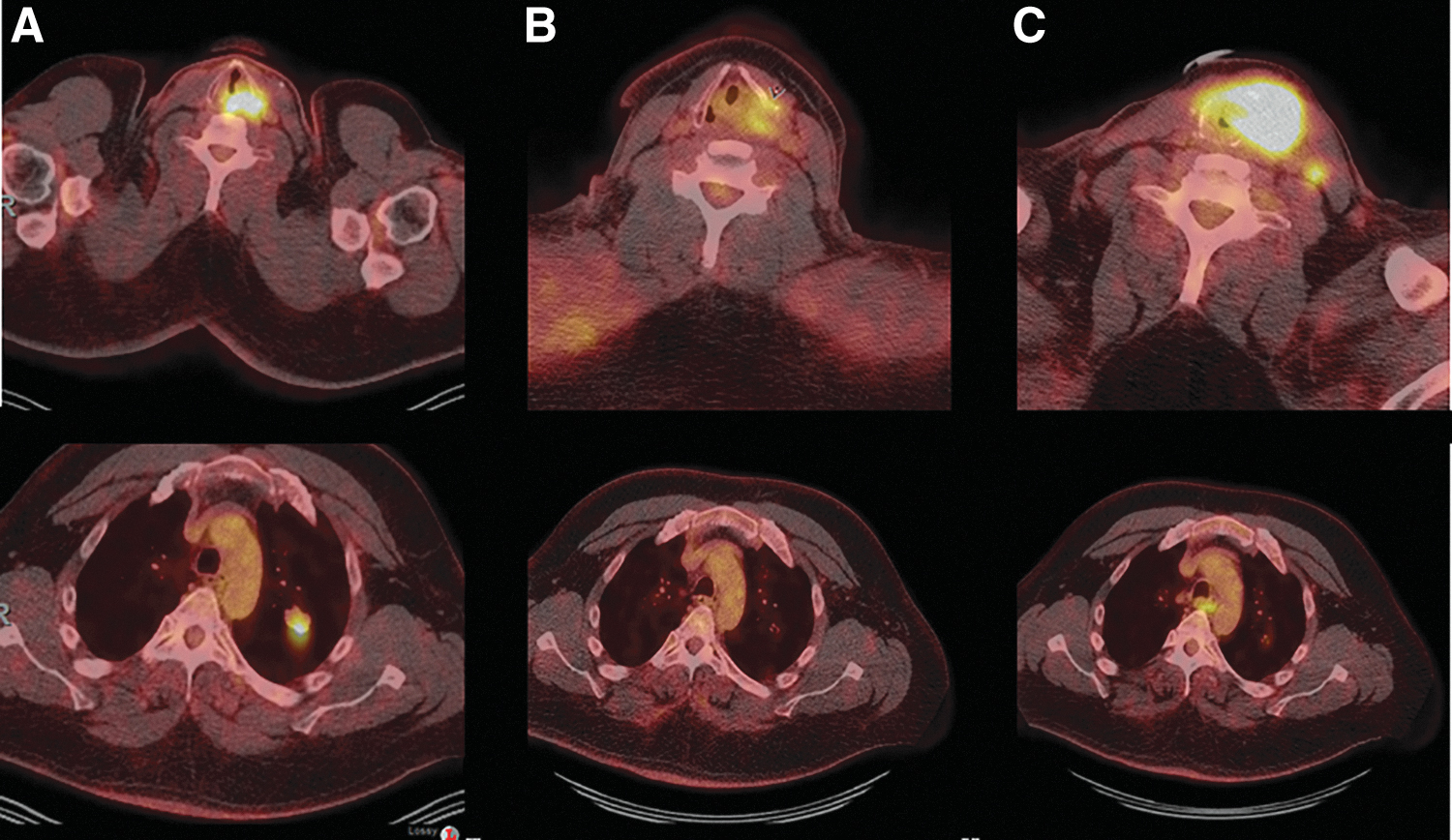
Patient 4's F18-FDG PET/CT axial image through the neck and chest at baseline (
Discussion
BRAF-targeted therapies have been shown to be effective in BRAF-mutated PTC (12) and ATC (16,17), and dabrafenib in combination with trametinib is FDA approved for the latter indication. However, development of resistance to kinase inhibitors presents a significant clinical challenge in the management of solid tumors. In this article, we presented four patients with BRAFV600E-mutated thyroid cancers who progressed while on BRAF inhibitors. Patient 1 and 3 acquired a KRASG12V mutation in the progressive tumor, while patient 2 acquired an NRASQ61K mutation in a progressive lymph node, and patient 4 acquired an NRASG13D mutation as demonstrated by liquid biopsy performed at the time of radiographic disease progression.
Until recently, mutations in the RAS and BRAF genes were believed to be mutually exclusive, including in thyroid cancers and melanoma (36 –38), but other reports found concomitant mutations in rare instances, mainly in advanced stages of the disease (39 –41). These double events were attributed to heterogeneity of the tumors and emergence of specific tumor cell clones through selective pressure imposed by drug therapy. Data from Raaijmakers et al. demonstrated that NRAS gene mutations can co-occur with BRAF mutations in single melanoma cells during vemurafenib treatment (42). Similarly, a thyroid cancer cell line was shown to harbor both BRAFV600E and KRASG12D mutations after 5 months of exposure to vemurafenib, and comparison of allelic frequency with that of untreated cells using ddPCR demonstrated that the KRASG12D mutation was acquired during treatment (33). In addition, a clone of cells harboring both the BRAFV600E and KRASG12D mutations was isolated from this population, demonstrating that, similar to melanoma cells, mutations in the BRAF and RAS genes are not mutually exclusive in single PTC cells (43). Therefore, while genetic heterogeneity of the primary tumor and selective pressure provided by the drugs will favor the emergence of a more aggressive tumor cell sub-population, acquisition of additional driver mutations in single tumor cells during therapy will further accelerate clonal evolution.
Expanding our understanding of mechanisms of acquired secondary resistance in BRAF-mutated thyroid cancer cells is vital to improving patient outcomes. Although resistance mechanisms to BRAF inhibitors have been described in other tumors including melanoma, lung adenocarcinoma, and colon cancer patients, events occurring in thyroid cancers might be distinct due to inherent transcriptional and epigenetic differences, and tissue-specific exposure to certain carcinogens.
In melanoma patients who were treated with vemurafenib or dabrafenib, comparison of whole-exome sequencing data from biopsies obtained at baseline and on progression revealed genetic alterations in ∼51–58% of the cases (44). These genetic alterations often reactivate the MAPK pathway and are most often detected in the NRAS gene (17–20%). These include NRASQ61K/L (21,42,44 –46), NRAST58 (44), and NRASG12/13 (46). Secondary resistance mechanisms through KRAS mutations (KRASG12D and KRASK117N) exist in melanoma patients but are rare, and mainly found in xenografts or cell lines (46,47). Other mutations conferring secondary resistance in melanoma are mainly found in the MEK1/2 (13%) and PTEN genes (11%) (44,46).
BRAF-mutated colorectal cancers respond poorly to vemurafenib and dabrafenib, which is possibly due to adaptive feedback reactivation of MAPK signaling mediated by EGFR (48,49). Other acquired resistance to kinase inhibitors are due to wild-type KRAS amplifications (50), RAF dimerization (51), and mutations in the KRAS gene (KRASG12R) (52). Results of a clinical trial combining BRAF and EGFR inhibition with or without MEK inhibition in BRAFV600E-mutated colorectal cancers indicated that 14 out of 29 evaluable patients (48%) developed ≥1 detectable KRAS or NRAS mutations in cfDNA at the time of disease progression, which were not detectable at baseline. Emerging mutations were KRASG12D, KRASG12C, KRASG12V, KRASG12A, KRASG13D, and NRASQ61L (53).
The BRAFV600E mutation is found in ∼2% of lung adenocarcinomas (54), and mechanisms of resistance to BRAF inhibitors in these tumors have only recently been reported. In one case study, Rudin et al. described emergence of the KRASG12D mutation along with a TP53 mutation on treatment with dabrafenib (55). In another study, resistance to vemurafenib could be overcome with the combination dabrafenib/trametinib, but the acquired mutation(s) were not described (56). A third study identified acquired NRASQ61K as a possible cause of dabrafenib plus trametinib resistance (57). Altogether, these data combined with ours confirm that KRASG12 is a hotspot mutation emerging in secondary resistance in advanced colorectal, lung, and thyroid carcinomas, but not in melanomas, while NRASQ61K is found in all four of these tumor types.
In the present article, we describe the emergence of RAS mutations such as KRASG12V, NRASQ61K, and NRASG13D as mechanisms of resistance in PTC and ATC. Owen et al. previously published their case of emergent KRASG12V mutation at the time of progression on dabrafenib plus trametinib (34). Other proposed mechanisms of acquired resistance in BRAF-mutated thyroid cancers have included epigenetic PI3K/AKT pathway activation through autocrine NRG1-mediated HER3 receptors (30), and autocrine IL-6-mediated JAK/STAT3 pathway activation (58). Duquette et al. reported that coexistence of BRAFV600E with either gain of the survival factor MCL1, or loss of the tumor suppressor P16, is associated with primary resistance to vemurafenib in patients with metastatic PTC (31). Further, we recently demonstrated the coexistence of both a KRASG12D and BRAFV600E mutation in a PDTC cell line, which was associated with cellular dedifferentiation and increase of rate of proliferation (33). The KRASG12D mutation emerged during long-term treatment with vemurafenib and coincided with resistance to the drug. Interestingly, treating the cells with an MEK inhibitor together with vemurafenib had less of an effect on proliferation than adding a PI3K inhibitor (33). This is attributed to the fact that these cells rather used the PI3K/AKT pathway, as shown by a significant increase of phosphorylated AKT over phosphorylated extracellular receptor kinase (ERK). Earlier mutagenesis studies have shown that specific RAS mutations may have distinct functional consequences on cell growth (59,60), and evidence now suggests that specific amino acid substitutions at an RAS hotspot have distinct consequences on effector signaling. For example, Ihle et al. recently compared downstream signaling in KRASG12C, KRASG12V, and KRASG12D-mutated nonsmall cell lung cancer (NSCLC) patients and cell lines. They showed increased AKT phosphorylation downstream of KRASG12D in comparison to KRASG12C/V that specifically activated RAS-like signaling (61). Thus, similar to the situation in NSCLC, the type of KRAS G12 substitution (C vs. D vs. V) occurring in thyroid cancers may drive a specific pathway, which has implications for which drug to introduce at progression, particularly as there is a drug being studied in the clinic that targets KRASG12C mutations (62).
The MEK inhibitor trametinib is approved in combination therapy with dabrafenib for the treatment of BRAFV600E melanoma. BRAF amplifications, BRAF splice variants, and epigenetic alterations all reactivate the ERK1/2 pathway in presence of BRAF inhibitors alone. However, despite its ability to inhibit proliferation of BRAFV600E-mutated melanoma cells or RAS-mutated RET/PTC thyroid cancer cells (29), our study shows that trametinib can be ineffective against emergence of RAS mutations (patients 2, 3 and 4). In melanoma, resistance to BRAF or MEK inhibitors is associated with the induction or persistence of activity of the AKT pathway in the presence of these drugs (63). Mechanisms of resistance to MEK inhibitors in colorectal cancer include alternative MEK1/2 activators such as MAP3K8 (64), high expression of inflammation-related genes (65), and emerging mutations in MEK1 that either prevent drug binding (24) or activate the gene (25). Mining of TCGA through cBioportal (66) indicated that only 4% of BRAFV600E PTC patient samples had elevated expression of inflammation-related genes such as IFIT1 and MX1, and only a few samples harbored elevated MEK1/2 or MAP3K8 expression. No MEK mutations could be detected by NGS in the samples from the patients at progression described in this study. Therefore the mechanisms of resistance to MEK inhibitors in thyroid cancers need to be further investigated.
Summary and Conclusion
We report the emergence of novel KRAS and NRAS mutations in four BRAFV600E-mutated thyroid cancer patients treated with BRAF inhibitors, and suspect that these mutations represent secondary mechanisms of BRAF inhibitor resistance. At this time, the clinical data available are insufficient to make treatment recommendations for these patients, but the type of KRAS amino acid substitution might have an impact on downstream signaling in these tumor cells. Therefore, possible ways to overcome resistance at the present time include the use of selective inhibitors of KRASG12C, PI3K inhibitors, ERK inhibitors, or immunotherapy combined with BRAF inhibitors. In the future, detailed characterization of the molecular changes in progressive thyroid tumors will point to further therapeutic opportunities, and may identify novel approaches to prevent emergence of drug resistance.
Footnotes
Author Disclosure Statement
M.E.C. has Merck and Genentech research funding, R.D. has Merck research funding, N.L.B. has Novartis research funding, and M.Z. has Merck research funding.
Funding Information
No funding was received.