
Abstract
Background:
Sporadic medullary thyroid carcinoma (MTC) is a relatively uncommon neuroendocrine malignancy and the molecular tumorigenesis of its sporadic type (sMTC) is only partially understood. In this study, we performed a study focusing on the genomic and transcriptomic characterization of sMTC.
Methods:
Twenty-nine sMTC patients were included. Whole-exome sequencing (WES) was carried out in 18 patients, including both tumor samples and matched noncancerous tissues. Whole transcriptome sequencing (RNA-Seq) was performed in all 29 tumors. WES, RNA-Seq, and copy number alteration (CNA) data were analyzed. A Cell Counting Kit-8 (CCK-8) assay was used to evaluate cell proliferation.
Results:
Among the somatic mutations, RET was the only recurrently cancer-related mutated gene (5/18, 27.8%). In the germline, FAT1 and FAT4, two members of the FAT gene family, were identified as the two most common mutated genes. CNA analysis found that FAT1 and FAT4, both located on chromosome 4q, were also two of the genes most commonly affected by somatic chromosomal deletions (4/18, 22.2%). Using TT and MZ-CRC-1 cell lines, the CCK-8 assay showed that FAT1 and FAT4 knockdown could promote MTC cell proliferation. Based on the gene expression profile, patients were clustered into two molecular subtypes: the mesenchymal-like subtype is characterized by epithelial–mesenchymal transition, while the proliferative-like subtype is associated with enrichment of cell cycle pathways. Most events of structural recurrence (80%) occurred in the proliferative-like subtype.
Conclusion:
In addition to RET, these findings demonstrate that FAT1/FAT4 genomic alterations appear to be frequent in sMTC. Two molecular subtypes of sMTC with distinct biological behavior could be identified. However, these results need to be validated by larger samples and more comprehensive experiments in the future, especially for the frequency and function of FAT1/FAT4 germline variants.
Introduction
Medullary thyroid carcinoma (MTC) is a relatively rare neuroendocrine malignant tumor arising from calcitonin-producing parafollicular cells (C-cells). MTC accounts for 3–5% of all thyroid malignancies, but is responsible for nearly 13% of thyroid cancer-associated deaths (1,2). Compared with well-differentiated thyroid carcinoma, MTC is biologically more aggressive, with an increased likelihood of widespread nodal metastasis and locally advanced invasion. Furthermore, MTC does not respond to radioactive iodine or thyroid-stimulating hormone suppression and is also insensitive to radiation therapy and conventional cytotoxic chemotherapy, leading to limited therapeutics in the setting of advanced disease.
About 75–80% of MTCs present as sporadic tumors (sMTCs) and the remaining 20–25% in a hereditary form (3,4). RET proto-oncogene mutations have been confirmed as the oncogenic drivers of nearly all hereditary MTCs and ∼50% of sporadic MTCs. Sporadic disease lacking somatic RET mutations may also bear mutations in RAS family members (5). However, the gap in our understanding of genetic events in a proportion of sMTCs prevents us from identifying patient-specific tumorigenesis and moving toward a more precise treatment for all patients. Therefore, additional genomic studies are required to better understand the tumorigenesis of sMTCs.
To the best of our knowledge, only a handful of high-throughput sequencing studies of MTC have been reported (6 –8) and few novel findings have been identified, indicating a gap of knowledge in the molecular signatures of this disease. Therefore, we performed a study characterizing genomic and transcriptomic profiles of sMTCs using whole-exome sequencing (WES), whole transcriptome sequencing (RNA-Seq), and copy number alteration (CNA) analysis.
Materials and Methods
Patient samples and clinicopathologic information
The study cohort comprised 29 sMTC patients with fresh-frozen, primary tumor samples, among whom 18 patients (MTC-2, -7, -8, -10, -14, -20, -21, -22, -23, -25, -26, -27, -29, -30, -31, -34, -38, and -39) had matched, fresh-frozen, noncancerous thyroid tissues. The matched, noncancerous thyroid samples were dissected more than 1 cm away from the tumor (either in the ipsilateral or contralateral lobe) and they were all microscopically confirmed to be free of cancer involvement by multiple frozen sections. All samples were obtained from the Tissue Bank of Fudan University Shanghai Cancer Center (FUSCC) after approval of the research by the Institutional Review Board, and written informed consent was obtained from each patient for sample collection. All the 29 sMTC patients underwent initial surgical treatment at FUSCC between 2007 and 2018 with a median follow-up period of 53 months (range: 2–108 months). During the follow-up period, five patients (17.2%) developed structural recurrence confirmed by fine-needle aspiration biopsy or repeated surgery; one patient (MTC-9) had mediastinal lymph node metastasis, while four patients (MTC-1, -11, -21, and -25) had cervical node metastases. Additional clinicopathologic and follow-up information is listed in Supplementary Table S1.
Whole-exome sequencing
WES was performed on 18 patients using DNA from both tumor samples and matched, noncancerous thyroid tissues. Genomic DNA was extracted using the AllPrep DNA/RNA Mini Kit (Qiagen, German). Genomic DNA (200 ng) from each individual was sheared using the Biorupter (Diagenode, Belgium) to acquire 200–300-bp fragments. The ends of DNA fragments were repaired and the Illumina adaptor was added (Fast Library Prep Kit, iGeneTech, Beijing, China). After the sequencing library was constructed, whole exomes were captured with the AIwholeExomeCNV Enrichment Kit with copy number variation (CNV) probes (iGeneTech, Beijing, China) and sequenced on an Illumina platform (Illumina, San Diego, CA) with 150-bp paired-end reads.
Alignment and variant calling
Raw reads were filtered to remove low quality reads using FastQC. Clean reads were mapped to the reference genome GRCh37 using BWA. After removing duplications, the single-nucleotide variant (SNV) and insertion and deletion (Indel) were called and annotated using the Genome Analysis Toolkit (GATK) based on dbSNP build 150 (9). All variants were annotated with ANNOVAR (10). A series of filtering criteria were applied to the variant candidates to finally identify SNVs and Indels: (1) variants with a quality <30, a depth of coverage <20 in tumor or plasma samples, or <2 reads supporting the variant were filtered out; (2) only variants within the exonic or splicing site were retained; (3) variants reported in more than 1% of the population in the 1000 Genomes Project (1000gAUG_2015ALL) or the Exome Aggregation Consortium (ExAC_ALL) or the Genome Aggregation Database (gnomAD_ALL) in the East Asian population were discarded as they were regarded as single-nucleotide polymorphisms (11,12); and (4) synonymous variants were excluded.
Identification of somatic cancer-related mutations and germline mutations
For somatic mutations, we used similar in silico methods in previous studies to identify cancer-related variants, including how to determine the harmfulness of variants and annotated SNVs (13 –15). Several functional prediction algorithms were used to evaluate the effect of each variant, including SIFT (16), Polyphen2 (17), and MutationTaster (18), with default settings. Only variants that were predicted to be deleterious, damaging, or disease by at least one tool were retained. Indels annotated in the Catalogue of Somatic Mutations in Cancer (COSMIC v70) database were included (19). To assess cancer-related variants, we collected four gene sets that were associated with cancer development and progress in previous studies, spanning 3823 unique genes (20 –23).
For germline mutations, using the InterVar database (24), all variants were classified following the guidelines of the American College of Medical Genetics and Genomics (ACMG) (25), which are commonly used for the interpretation of germline variants. Similar to a previous study (26), only germline mutations classified as pathogenic, likely pathogenic, or variants of uncertain significance (VUS) based on the ACMG guidelines were retained in our study.
CNA calling
CNVkit was implemented with default parameters on WES data (27). Due to the inherent limitation of CNV measurement using NGS data, we limited our analysis to arm-level alterations, which were defined as those spanning >50% of a chromosome arm with |log2 ratio| > 0.2 (28). Chromosome cytoBand was downloaded from the UCSC browser and bedtools intersect was used to annotate the arm-level segment. The cutoff (|log2 ratio| > 0.2) was also used to select cancer genes affected by arm level. The
Pathway-associated cancer gene mutations
We used the integrated oncogenic signaling pathways from previous studies (29), including cell cycle, HIPPO, MYC, Notch, NRF2, PI3K, TGF-Beta, receptor tyrosine kinase (RTK)-RAS, TP53, and Wnt. Using all somatic cancer-related mutated genes or CNV-involved genes, we calculated the enrichment p value for each oncogenic signaling pathway using the R package, clusterProfiler, with default parameters (30). Visualization of the highlighted pathways was completed by the PathwayMapper tool (31).
Cell culture and reagents
Two commonly used MTC cell lines, TT and MZ-CRC-1, were used for the experiment in our study. Both cell lines were provided by the Cell Bank of University of Colorado Cancer Center. TT cells were cultured in Ham's F-12K (Kaighn's) medium (Gibco, Grand Island, NY), while MZ-CRC-1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco). Both media were supplemented with 10% fetal bovine serum (Gibco), 1% penicillin, and 1% streptomycin.
Small interfering RNA transfection and Cell Counting Kit-8 assay
Small interfering RNAs (siRNAs) against FAT1 and FAT4 and corresponding negative controls were synthesized by RiboBio Biotechnology (Guangzhou, China). Knockdown efficiency was evaluated by quantitative real-time polymerase chain reaction (RT-qPCR) after 48 hours of transfection. For the two genes tested, expression was affected by two siRNAs with completely unrelated sequences. The siRNA targeting sequences of the FAT1 gene were GGACCGAAATTCCTTCGAA (siFAT1-1) and GGACGTGCCTATTGGAACA (siFAT1-2), respectively, while the siRNA targeting sequences of the FAT4 gene were CTACGAAGATTGGCTGTGA (siFAT4-1) and GACGCCAAGTCATCTTAGA (siFAT4-2), respectively. The cells were transfected at a final concentration of 20 nM using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's instructions.
The cells were seeded into 96-well culture plates with 2 × 103 cells/well. The cells were transfected after 24 hours of incubation. Subsequently, 10 μL of Cell Counting Kit-8 (CCK-8) solution (Dojindo Laboratories, Kumamoto, Japan) was added in 90 μL of corresponding medium (F-12K for TT and DMEM for MZ-CRC-1). The plates were then incubated for 2 hours, and the optical density at 450 nm (OD450) was measured using an automatic microplate reader (Synergy 4; BioTek, Winooski, VT). Similar to a previous study (32), although the cells were transiently transfected, their OD450 values were continuously measured for five days because of the more prolonged doubling times of the two MTC cell lines. Three independent experiments were performed in triplicate.
RNA-Seq
Tissues used for RNA isolation were stored in RNAlater stabilization solution (Thermo Fisher Scientific, Waltham, MA). RNA was isolated using an AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. The RNA quality was measured with an Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). All 29 sMTC samples had an RNA integrity number higher than 7.0 and were further processed for RNA-Seq. Approximately 1 μg of total RNA was treated with the Ribo-off rRNA Depletion Kit (Vazyme, Nanjing, China) before constructing the RNA-Seq libraries. RNA-Seq libraries were prepared using the VAHTS Total RNA-Seq (H/M/R) Library Prep Kit for Illumina (Vazyme) following the manufacturer's instructions. Briefly, ribosome-depleted RNA samples (∼100 ng) were fragmented and used for first- and second-strand cDNA synthesis with random hexamer primers. cDNA fragments were treated with the DNA End Repair Kit to repair the ends and modified with Klenow to add an A at the 3′ end of the DNA fragments; the fragments were then ligated to adapters. Purified dsDNA was subjected to 12 cycles of PCR amplification, and the libraries were sequenced by an Illumina sequencing platform on a 150-bp paired-end run.
Quantification of RNA expression levels
RNA-Seq reads were mapped onto the reference human genome GRCh38 using two-pass mapping of STAR (v2.5) (33,34). The number of reads per gene was counted with featureCounts using the GENCODE v29 annotation (35). Each gene expression level normalized to transcripts per million was computed using a custom PERL script.
Unsupervised hierarchical clustering
To perform gene expression clustering, we reduced the data to the top 1000 most variable genes, measured by median absolute using the R package, ConsensusClusterPlus. Before using default settings of the agglomerative hierarchical clustering algorithm with Pearson correlation distance to cluster samples, we normalized gene expression by median center (ConsensusClusterPlus [Tutorial]). A consensus cumulative distribution function (CDF) plot was used to determine the optimal number of clusters.
Survival analysis
Survival analysis was performed using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA) with both the log-rank test and Wilcoxon test.
Pathway enrichment analysis
Gene set enrichment analysis (GSEA) for the cancer hallmark signatures (MSigDB 6.0) between the two clusters was performed (36). Fold changes between two clusters were log2 transformed and used to generate a ranked gene list, and then preranked GSEA was used to determine pathway enrichment.
Results
Mutations identified by WES
Eighteen tumor samples and their paired noncancerous thyroid tissues were included in the WES analysis. For tumor samples, the mean sequencing depth was 120 × , with at least a 20 × depth in 97.08% of the target exome regions, while for noncancerous tissues, the mean sequencing depth was 150 × , with at least a 20 × depth in 97.84% of the target exome regions (Supplementary Table S2).
A total of 48 somatic cancer-related mutations were detected, including 45 missense substitutions, 1 nonsense mutation, and 2 nonframeshift deletions (Fig. 1A and Supplementary Table S3). The 18 patients harbored only 2.7 somatic cancer-related mutations per tumor, which was comparable with a very low burden (13 somatic cancer-related mutations in 7 sporadic MTCs, average: 1.86 per tumor) reported in a previous study (8).
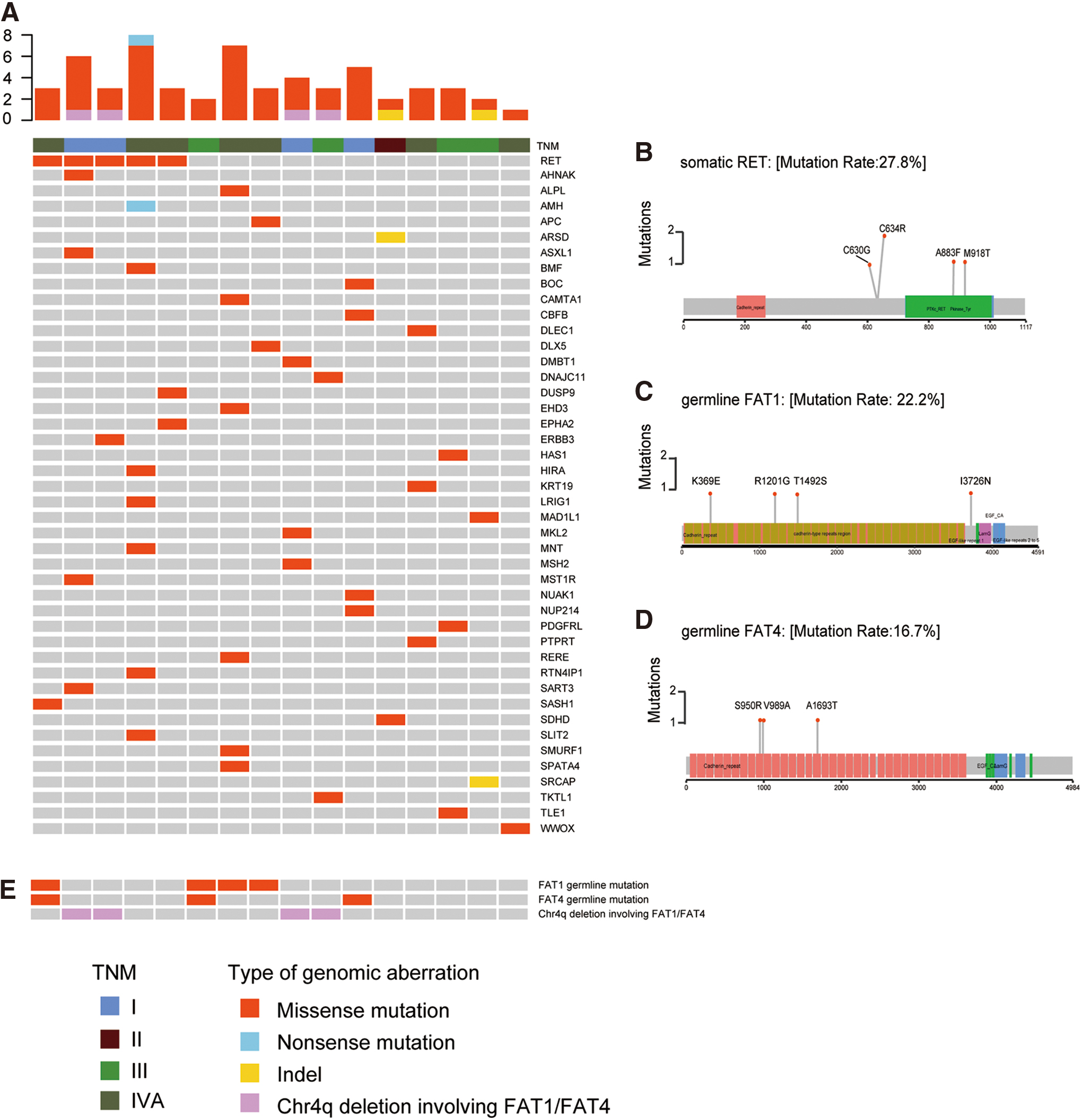
The mutational characterization of sMTC. (
RET (n = 5, 27.8%) was identified as the only gene with recurrent somatic mutations (mutated in ≥2 cases), including p.C634R (n = 2), p.M918T (n = 1), p.A883F (n = 1), and p.C630G (n = 1) (Fig. 1B and Supplementary Table S3). However, we did not detect any somatic cancer-related mutations in three tumors (MTC-T2, -T34, and -T38). In addition, no somatic RAS mutation was found in these 18 patients.
Furthermore, 203 germline mutations classified as VUS, pathogenic, or likely pathogenic based on the ACMG guidelines were identified. We found that FAT1 (n = 4) and FAT4 (n = 3), two tumor suppressor genes belonging to the same atypical cadherin superfamily, were most commonly mutated in the germline (Supplementary Table S4 and Fig. 1C–E). All FAT1 and FAT4 germline variants were VUS.
All the RET, FAT1, and FAT4 mutations were validated by Sanger sequencing.
CNA analysis
For the 18 patients with tumors and matched noncancerous tissues, CNA analysis was further conducted to assess chromosome instability in MTC. No germline CNVs were found in the CNA analysis. On the other hand, 31 somatic arm-level CNVs involving 754 reported oncogenes and tumor suppressor genes were detected in 9 (50.0%) tumor samples (Supplementary Tables S5–S7). The most frequent somatic changes included copy number losses in chromosome 4q (n = 4, 22.2%) and 1p (n = 4, 22.2%) (Supplementary Table S5). The heatmap of somatic CNVs is shown in Figure 2A. Comparison of chromosomal stability between tumors and matched noncancerous tissues is listed in Figure 2B–D and Supplementary Figure S1.
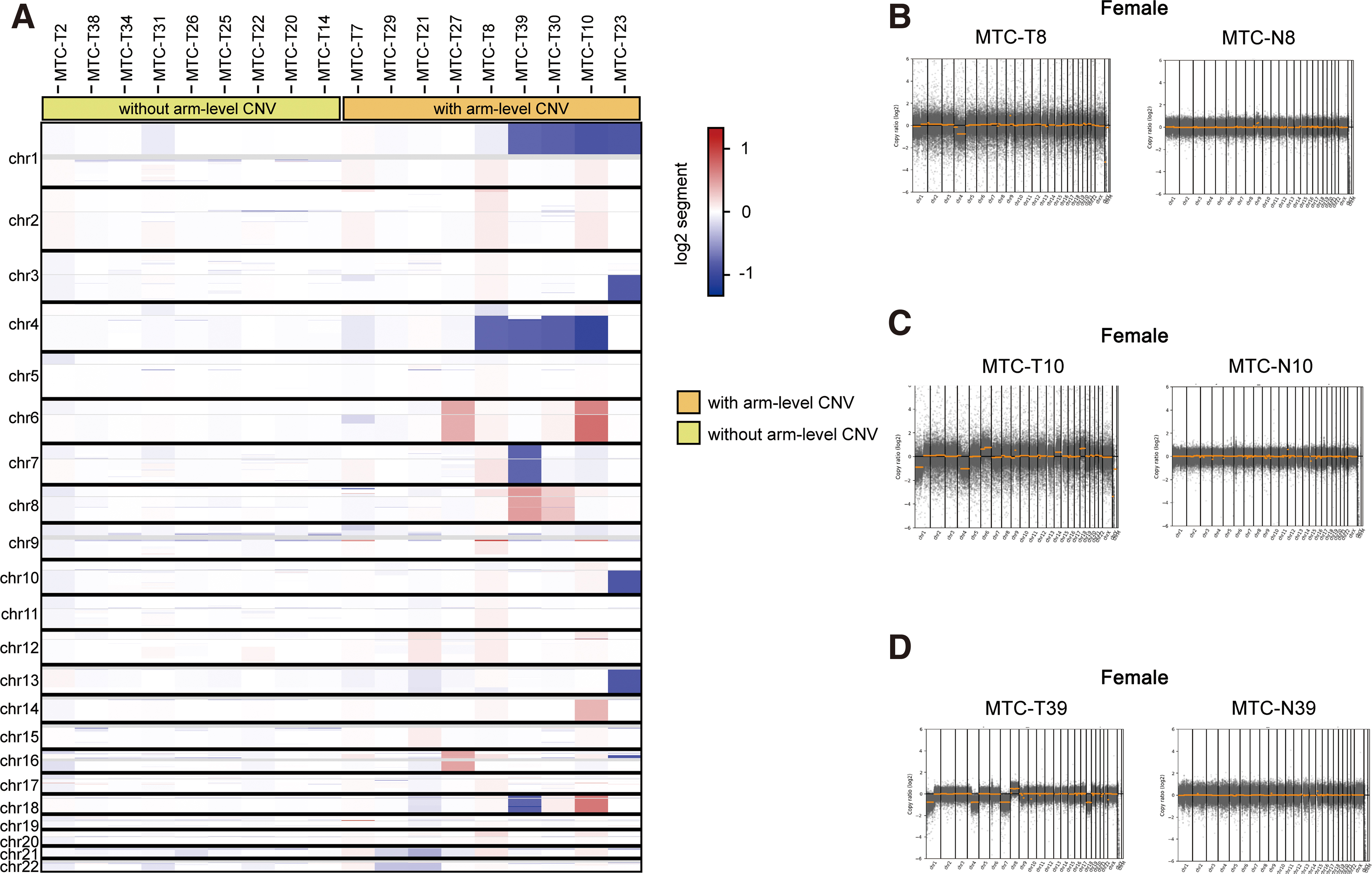
(
It is noteworthy that FAT1 and FAT4 (both located on chromosome 4q) were still two of the genes most frequently affected by these large-fragment, somatic chromosomal changes (n = 4) (Supplementary Table S7 and Fig. 1E).
Integrating the results of WES and related CNA analysis, we found that the somatic genomic events were highly concentrated in members of the RTK/RAS pathway (including RET, ERBB3, and the RasGAP gene family and others) and the Hippo pathway (including FAT1, FAT4, LATS2, and others) (Fig. 3). Overall, 50.0% (n = 9) of MTC samples displayed somatic SNVs or CNVs in the RTK/RAS signaling axis, while 44.4% (n = 8) displayed alterations in genes of the Hippo pathway. Six samples (33.3%, MTC-T8, -T10, -T23, -T27, -T30, and -T39) demonstrated somatic SNVs or CNVs in genes of both pathways (Supplementary Table S8).
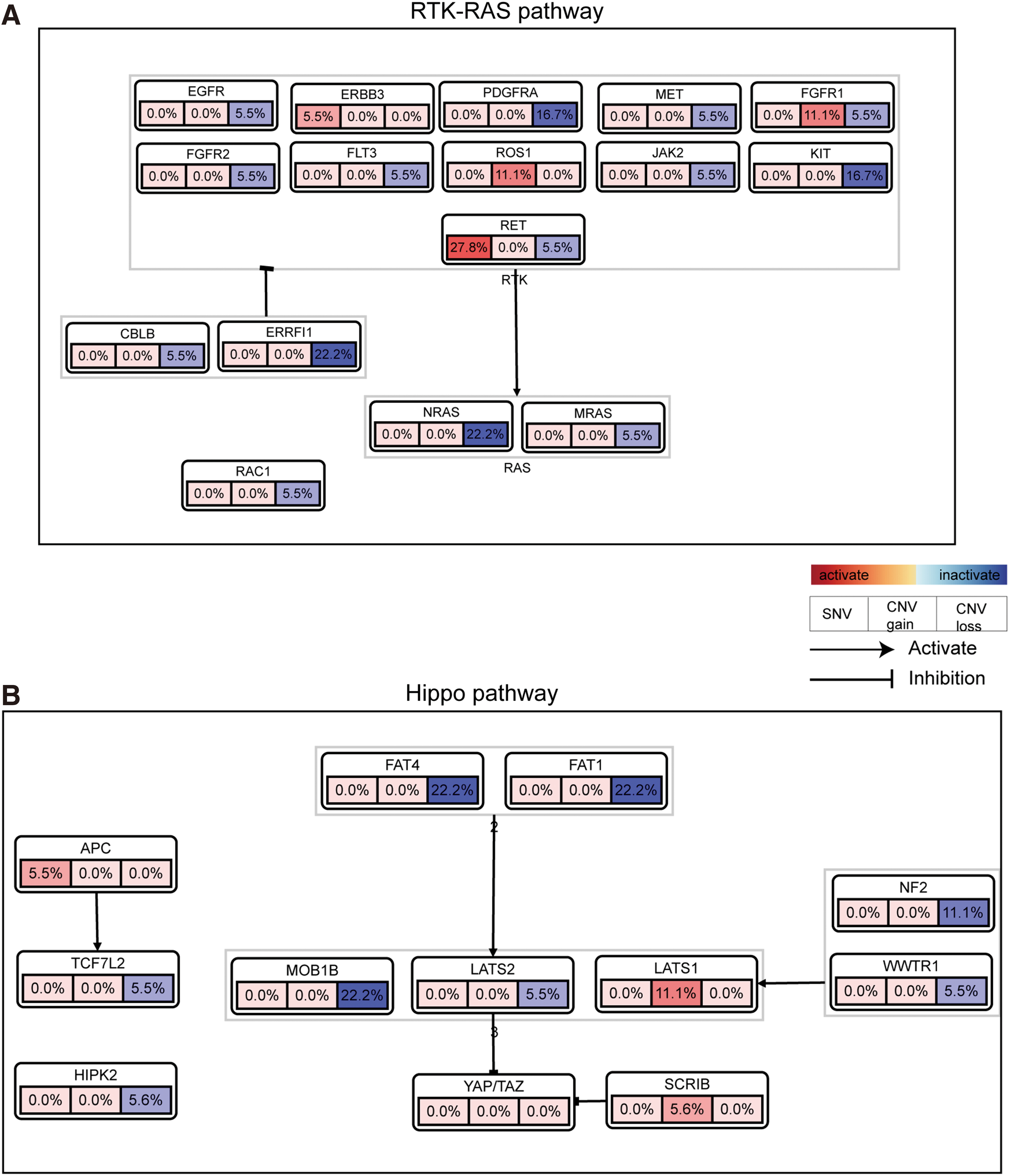
Somatic genomic alterations in genes of the (
CCK-8 assay
Using TT and MZ-CRC-1 cell lines, the CCK-8 assay was used to explore the effect of FAT1 and FAT4 knockdown on MTC cell proliferation. As mentioned above, similar to a previous study (32), although the cells were transiently transfected, their OD450 values were continuously measured for five days due to the prolonged doubling times of the two cell lines. The CCK-8 results showed that FAT1 and FAT4 knockdown could significantly promote cell proliferation in both TT and MZ-CRC-1 cells (Fig. 4).
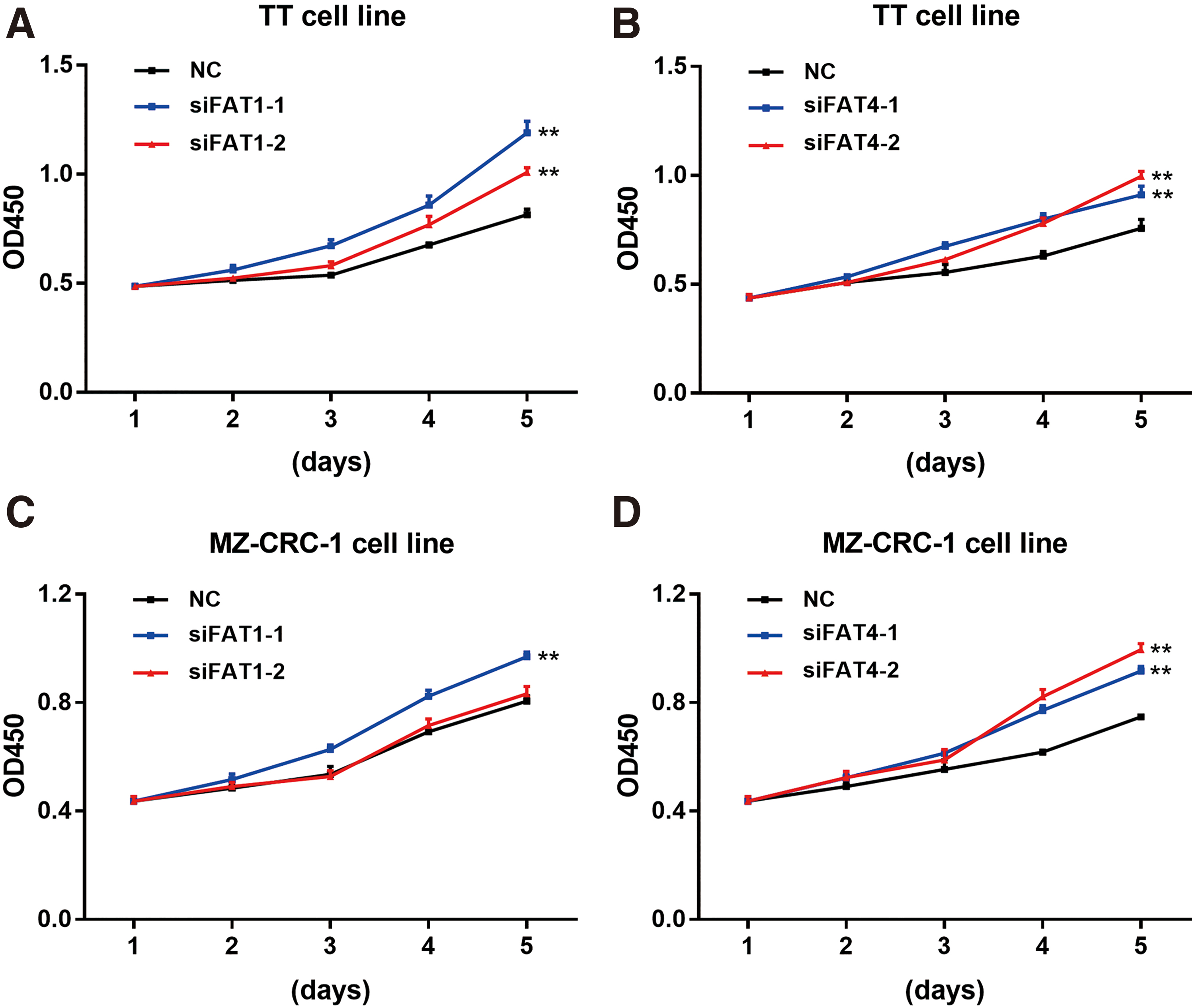
Cell proliferation curves detected by CCK-8 assay. (
Molecular subtyping using RNA-Seq data
RNA-Seq was performed and analyzed in all 29 sMTC tumor samples. Unsupervised hierarchical clustering of RNA-Seq data divided the 29 samples into distinct subgroups. The optimal number of clusters was determined as two, as a peak of CDF statistics was observed at k = 2 (Supplementary Fig. S2 and Fig. 5A). GSEA was then performed to analyze the cancer hallmark signatures of the two clusters (Cluster 1 and Cluster 2). We found that the epithelial–mesenchymal transition (EMT) gene set was enriched in Cluster 1, while MYC targets V1 was enriched at a high degree in Cluster 2 (Fig. 5B–D). Therefore, consistent with previous studies of other cancers (37 –39), Cluster 1 and Cluster 2 were termed as the mesenchymal-like subtype and proliferative-like subtype, which were characterized by EMT and enhanced proliferative activity, respectively. In addition, GSEA also demonstrated other profile differences between the two clusters. TNFA signaling through NFKB, inflammatory response, allograft rejection, and hypoxia gene sets were enriched in the mesenchymal-like subtype, while the proliferative-like subtype showed upregulation of protein secretion, oxidative phosphorylation, and DNA repair gene sets (Fig. 5B).
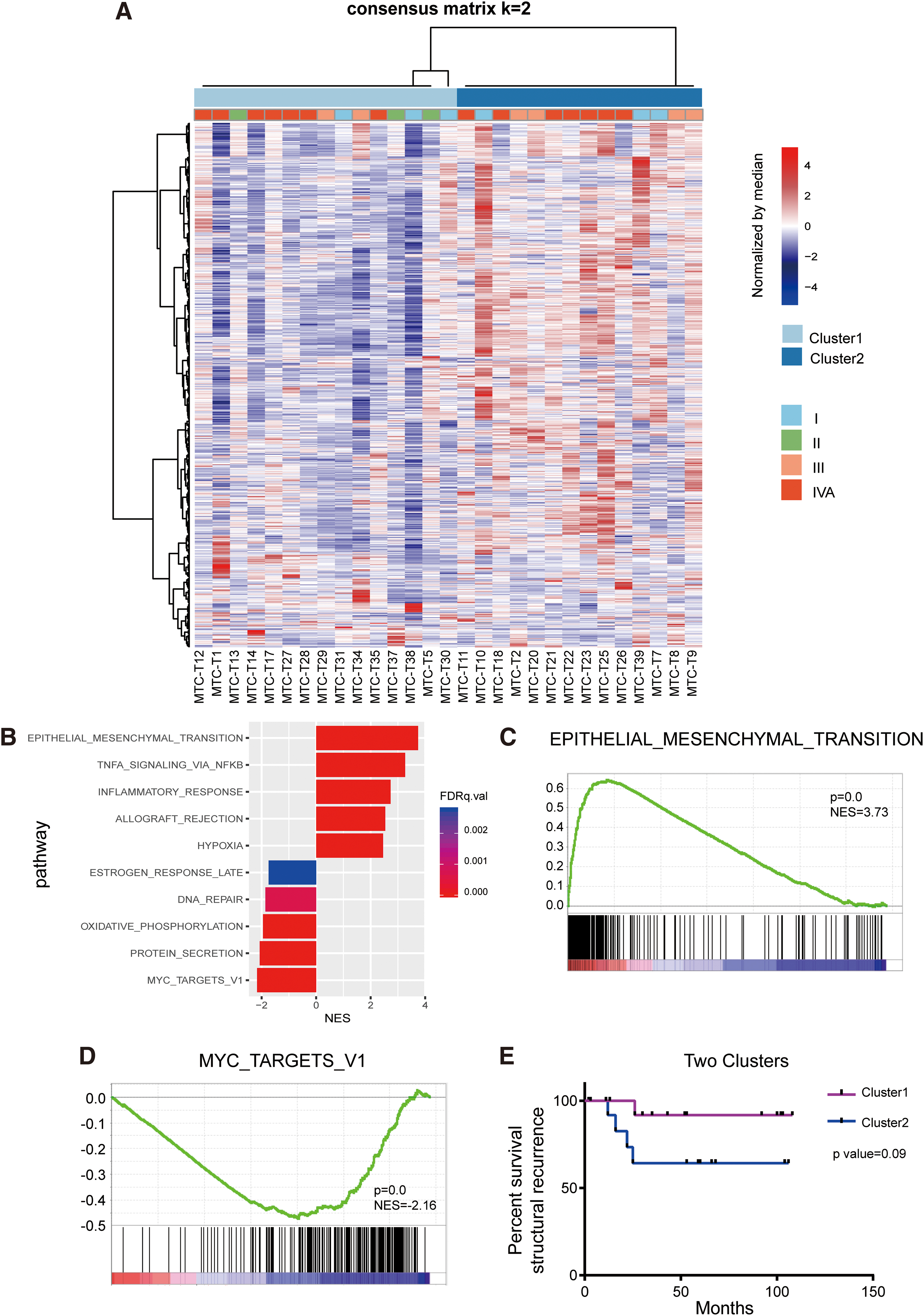
Molecular subtyping based on RNA-Seq data. (
We further assessed the clinical relevance of the two molecular subtypes. Of the 15 patients with the mesenchymal-like subtype (Cluster 1), only 1 (6.7%) developed structural recurrence during the follow-up period, while most structural recurrence events (4/5, 80%) occurred among the 14 patients with the proliferative-like subtype (Cluster 2), although the log-rank test failed to show a significant prognostic difference between the two subtypes (p = 0.09) (Fig. 5E).
Discussion
The understanding of the molecular pathogenesis of sMTC remains incomplete, and the COSMIC database demonstrates that ∼35% of sMTCs have no established drivers. There is an unmet need for studies further characterizing genomic and transcriptomic alterations in these tumors. In the present study, we investigated mutational, copy number, and transcriptional features of sMTCs using WES and RNA-Seq technologies.
Unsurprisingly, our genome-based analysis confirms the important position of RET proto-oncogene mutations in the mutation spectrum of sMTC. Of note, p.M918T is generally regarded as the most prevalent somatic RET mutation in sMTCs (40). However, in our cohort, the RET p.C634R mutation (n = 2) was more frequent than the p.M918T variation (n = 1)—this event may simply be caused by the small sample size of the study.
In addition, except for RET, no other recurrent, somatic cancer-related mutations have been identified. Prior studies have reported that RAS alteration is another important driver mutually exclusive of RET mutations (41,42): the COSMIC database also shows that 10% and 3% of MTC patients harbor HRAS and KRAS mutations, respectively. However, no somatic RAS mutation was identified in the present study. Similarly, in a study by Agrawal et al., no RAS mutation was detected among 12 RET wild-type patients in the discovery cohort (7). In another study including 330 MTC patients, RAS mutation-positive cases only accounted for 4.8% (43). Therefore, although our study is limited in sample size, somatic RAS mutations may not be prevalent in sMTCs, and other genomic events may be involved.
It is believed that aberrations at the chromosomal level are closely related to tumorigenesis and disease progression. Previous studies have demonstrated frequent deletions of chromosome 1p in MTC (44 –47). Del(1p) is also one of the most common CNVs (n = 4, 22.2%) in our study. It has been reported that loss of CDKN2C, a known tumor suppressor gene located on chromosome 1p32, could act as a diagnostic and prognostic indicator in MTC (48,49). Similarly, we observed this chromosomal imbalance in all four patients with del(1p) in the current study. In addition, del(4q) has also been observed as a common somatic genomic event (n = 4, 22.2%) in our study, a deletion that has also been reported in prior studies (44,45). Loss of chromosome 4q is commonly observed and regarded as a probable cause in numerous cancers (50 –53). Interestingly, the two most common germline mutated genes in the WES analysis presented here, FAT1 and FAT4, are both located on this chromosomal arm (FAT1:4q35; FAT4:4q28) and both genes are affected in the four cases with del(4q) in our cohort. In addition, other CNVs described in previous studies (44 –46,54), such as somatic loss of 22q, 13q, 7q, and 3q, have also been identified in this study, indicating that multiple chromosomal abnormalities are potentially involved in sMTC tumorigenesis.
In this study, FAT1 and FAT4 are not only two of the most common genes affected by deleterious CNVs but also two of the most common germline mutated genes. Furthermore, SNVs and deleterious CNVs occur in a total of 50% (n = 9) of tumor samples. Although the high percentages of FAT1/FAT4 aberrations have not been reported in other studies and may vary among different study populations, these results still prompt us to devote more attention to these two genes. Both genes encode transmembrane proteins belonging to the atypical cadherin superfamily that act upstream of the Hippo signaling pathway. FAT1 and FAT4 have been implicated to act as tumor suppressor genes in different human cancers. FAT1 inactivation has been reported to be correlated with the onset of glioblastoma multiforme, head and neck, and colon cancers (55). Deleterious FAT4 aberrations are commonly observed in melanoma, pancreatic, and gastric cancers (56). We therefore hypothesize that FAT1 and FAT4 also play a similar role as tumor suppressor genes in sMTC. The CCK-8 assay shows that FAT1 and FAT4 knockdown may promote MTC cell proliferation in vitro. However, in the future, more functional experiments and exploration of the underlying mechanism are required, especially for the FAT1 and FAT4 germline VUS.
The present study also represents the first demonstration of substantial tumor heterogeneity based on the gene expression profile and divides patients into two well-defined molecular subtypes: mesenchymal-like and proliferative-like groups. As the name suggests, the mesenchymal-like subtype is characterized by higher expression of EMT signature genes involved in extracellular matrix maintenance and cell-to-cell adhesion processes. Similar to a previous study on gastric cancer (57), the mesenchymal-like subtype is also associated with upregulated TGF-β pathway gene sets. Furthermore, an elevated inflammatory response and tumor hypoxia are also identified as hallmarks of this subtype. In contrast, enrichment of c-MYC target genes, activation of oxidative phosphorylation, and DNA repair are expected to increase the proliferative activity in the proliferative-like subtype. In terms of clinical relevance, most recurrences occur in the proliferative subtype, suggesting the possibility of a rapidly progressive clinical course in this subgroup of patients. Our findings suggest that MTC is genotypically heterogeneous and should not be regarded as a genetically monomorphus disease. Heterogeneity among tumors may result in distinct transcriptional features, prognoses, and even potential targets for treatment.
In summary, we report an analysis focusing on the genomic and transcriptomic characterization of sMTC. The findings show that FAT1/FAT4 genomic events may be frequent events in this rare neuroendocrine tumor. Identifying the distinct molecular subtypes may help to predict prognosis and may lead to improved therapeutic strategies. Our results warrant further validation with larger samples and more comprehensive experiments, especially for the frequency and function of FAT1/FAT4 germline variants.
Footnotes
Acknowledgments
The authors thank patients who have donated their specimen for this study. They also thank Liwen Bianji, Edanz Editing China, for editing the English text of a draft of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the Shanghai Municipal Health Bureau Project (No. 201840268).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8