
Abstract
Background:
Mutations of monocarboxylate transporter 8 (MCT8), a thyroid hormone (TH)-specific transmembrane transporter, cause a severe neurodevelopmental disorder, the Allan-Herndon-Dudley syndrome. In MCT8 deficiency, TH is not able to reach those areas of the brain where TH uptake depends on MCT8. Currently, therapeutic options for MCT8-deficient patients are missing, as TH treatment is not successful in improving neurological deficits. Available data on MCT8 protein and transcript levels indicate complex expression patterns in neural tissue depending on species, brain region, sex, and age. However, information on human MCT8 expression is still scattered and additional efforts are needed to map sites of MCT8 expression in neurovascular units and neural tissue. This is of importance because new therapeutic strategies for this disease are urgently needed.
Methods:
To identify regions and time windows of MCT8 expression, we used highly specific antibodies against MCT8 to perform immunofluorescence labeling of postnatal murine brains, adult human brain tissue, and human cerebral organoids.
Results:
Qualitative and quantitative analyses of murine brain samples revealed stable levels of MCT8 protein expression in endothelial cells of the blood
Conclusions:
With respect to MCT8-deficient conditions, our analyses not only strongly support the contention that the BBB presents a lifelong barrier to TH uptake but also highlight the need to decipher the TH transport role of MCT8 in early neuronal cell populations in more detail. Improving the understanding of the spatiotemporal expression in latter barriers will be critical for therapeutic strategies addressing MCT8 deficiency in the future.
Introduction
Thyroid hormone (TH) action is of critical importance for normal brain development and function. This is exemplified in children born with congenital hypothyroidism due to defects in thyroid gland development or function. These patients suffer from severe neurocognitive impairment, if not substituted early after birth with adequate doses of L-thyroxine (T4) (1). However, TH uptake into the highly shielded neurons in brain tissue is complex. The TH precursor T4 and the active TH 3,3′,5-triiodothyronine (T3) have to pass not only specialized endothelial cells of the blood
In this context, MCT8 is regarded to be of particular importance for TH uptake into the brain, as X-linked inactivating mutations of the SLC16A2 gene encoding MCT8 lead to a neurodevelopmental disorder with severe psychomotor impairment (9,10), known as MCT8 deficiency or Allan-Herndon-Dudley syndrome (11). Despite high serum concentrations of T3 (12), intracerebral TH concentrations are likely to be reduced in these patients, since histopathological findings of delayed myelination, synaptogenesis, and neuronal differentiation imply a cerebral hypothyroid state during critical periods of pre- and postnatal brain development (13,14). Importantly, trials to treat MCT8-deficient patients with TH analogues met with very limited success in improving neurological outcomes (15 –17). Only the early postnatal administration of the TH analogue Triac restored Purkinje cell morphology, myelination, and distribution of cortical interneurons in mouse models (18), and it slightly improved the neurological symptoms in six out of seven patients with MCT8 deficiency within the age group of 1.5 to 3.5 years (17,18). Therefore, the development of targeted therapies critically depends on (i) a detailed knowledge of the exact localization of MCT8 within the NVU to facilitate TH transport from the blood to neurons, and (ii) the identification of temporal changes of MCT8 expression to define the time window for therapy.
It is noteworthy that studies on the etiopathology of MCT8 deficiency in animal model systems are hampered by the existence of profound species-specific differences of TH transporter expression at the NVU level (3,19). Heuer et al. have first described pronounced Mct8 expression in TH-sensitive neuronal populations in postnatal murine brains (20). Although subsequent murine and human analyses have not revealed species differences of neuronal expression profiles (21 –27), single-cell RNA sequencing analyses suggest differential abundance of Mct8/MCT8 and other TH transporters (28). This is underscored by single Mct8 knockout (KO) mice that do not show overt neurodevelopmental defects but a few mild behavioral changes previously associated with either hypo- or hyperthyroidism (14,22). Although double KO mice for Mct8 and Oatp1c1 present defective TH transport at the NVU level and share some neurodevelopmental defects with human patients (29), the need for concomitant inactivation of two critical TH transporters to induce a phenotype complicates a direct translation of murine findings to the human situation. Since the development of novel treatment strategies and their validation in animal models will need a deeper understanding of the species-specific pathophysiological mechanisms of TH transport defects, detailed comparative expression studies of MCT8 will be needed to interrogate existing discrepancies among species.
Moreover, a large portion of available data on MCT8 expression is still limited to the description of transcript levels. Thus, there is a need to characterize protein abundance as a closer proxy for physiological relevance (30). This is particularly important given that previous studies showed dynamic changes in mRNA expression that are dependent on TH axis status (25,31 –33), sex (34), and fasting conditions (35,36), during critical illness (24,37) and throughout development (38,39). Studies investigating MCT8 protein expression are still limited due to a lack of specific antibodies and difficulties in obtaining human brain tissue for immunostaining (3).
In this study, we aimed at systematically elucidating the spatiotemporal pattern of MCT8 protein expression in human and murine neural tissue by using an optimized immunofluorescence staining protocol with highly specific anti-MCT8 antibodies. This work comprises serial sections of murine brains from young postnatal ages into adulthood, postmortem and postoperative brain tissue of adult human subjects, and cerebral organoids derived from human induced pluripotent stem cell (hiPSC).
Materials and Methods
Cells
For antibody validation, Madin-Darby canine kidney cell lines (MDCK1) were stably transfected with N-terminally hemagglutinin (HA)-tagged human MCT8 as described elsewhere (40). As MDCK1 cells did not express human MCT8 endogenously, cells stably transfected with an empty vector (pcDNA3) served as negative control. Cells were cultivated in Dulbecco's modified eagle medium (DMEM)/nutrient mixture F-12 (1:1), containing 5% fetal calf serum, 0.5% penicillin/streptavidin, and 200 μg geneticin 418/mL and seeded into six-well plates (4 × 105 cells/well). After 12 hours of incubation at 37°C and 5% CO2, medium was exchanged with 0.02 M phosphate buffered saline (PBS), pH 7.4. For protocol optimization, different fixatives were tested, including 100% ethanol (EtOH), 4% PBS-buffered paraformaldehyde (PFA), and acetone with short (10 seconds) versus long (20 minutes) incubation.
Murine tissue
Experiments involving animals were reported and performed in accordance to the ARRIVE guidelines, the European Union Directive 2010/63/EU, and the German guidelines for care and use of laboratory animals after approval by institutional authorities. Mice were kept under standard conditions (12 hours light/dark cycle, specific pathogen-free environment) with free access to food and water.
MCT8-KO mice were generated in C57BL/6 mice (Deltagen) by insertion of a lacZ-neomycin phosphotransferase-2 gene into exon 2 (14,22) and housed in the central animal facility of the Charité Berlin, Germany. Mice (KO n = 3, wildtype (WT) n = 6) were analyzed on postnatal day (P) 21 and P154.
In addition, systematic immunofluorescence analysis and semi-quantification of murine brain tissue was performed with wildtype C57BJ/6 mice that were raised and housed at the Medizinisch-Experimentelles Zentrum Leipzig, Germany, and they were sacrificed at P6, P12, P21, P83, and P155 (each n = 4).
At the indicated time points, brain tissue was removed, directly embedded in tissue freezing medium (Leica), and frozen in isopentane on dry ice. Serial coronal cryosections with 12-μm thickness were cut on a cryostat (Leica), and every fourth section was mounted on superfrost slides (Thermo Scientific) and stored at −20°C. Before immunofluorescence labeling, the tissue was fixed for 10 seconds with 100% EtOH.
Human tissue
Postmortem human brain tissue (including pituitary) of three body donors (one female, 82 years; two males, 78 and 82 years) with a postmortem delay of 20–48 hours was obtained from the Institute of Anatomy, Leipzig, Germany, after institutional approval for the use of postmortem tissues and in line with the Saxonian Death and Funeral Act of 1994, third section, paragraph 18, item 8. One fresh cortical postoperative brain sample was provided by the department of Neurosurgery (Charité, Universitätsmedizin Berlin, Germany) after approval by local authorities (EA2/111/14) and written informed consent of the subject (50-year-old woman with epilepsy) before the study. Brain regions of interest (ROIs) were removed, cryopreserved, and prepared for immunofluorescence labeling as described for murine samples.
Alternative tissue preparations were compared by fixing postmortem tissue in 4% PFA overnight at 4°C and embedding it in paraffin. For immunofluorescence labeling of paraffin-embedded samples, tissue sections were cut at 12-μm thickness on a microtome. After deparaffinization in xylene and rehydration, antigen retrieval was performed in 0.1 M citrate buffer (pH 6.0) at 95°C. For immunohistochemistry of tissue from PFA-fixed samples, sections were cut into slices of 35-μm thickness on a vibratome, stored in PBS, and mounted onto superfrost slides shortly before immunolabeling.
Human cerebral organoids
Cerebral organoids were generated from the human induced pluripotent stem cells (hiPSC) lines BIHi004-A and BIHi005-A by using a modified protocol of Lancaster and Knoblich (41). hiPSC were cultured in E8 medium (42) in Geltrex-coated culture plates (Thermo Fisher). On day 0 of organoid culture, hiPSC were dissociated by Accutase treatment to generate single cells. In total, 9000 cells were then plated in each well of an ultra-low-attachment round-bottom 96-well plate (Corning) in hPSC medium containing DMEM/F12 (Thermo Fischer) supplemented with 20% Knock Out Serum Replacer (Thermo Fischer), 3% embryonic stem cell-qualified fetal bovine serum (Millipore), Glutamax (Thermo Fisher), minimum essential media-nonessential amino acids (MEM-NEAA; Thermo Fisher), 2-mercaptoethanol (Thermo Fisher), 4 ng/mL basic fibroblast growth factor (bFGF; Peprotech), and 50 mM Rho-associated protein kinase (ROCK) inhibitor Y-27632 (Wako). Embryoid bodies were fed every other day for six days with hPSC medium, while bFGF and ROCK inhibitor were only supplemented until day 4. On day 7, medium was changed to neural induction medium containing DMEM/F12, 1 × N2 supplement, Glutamax, MEM-NEAA, and 0.18 U/mL heparin (Rotexmedica) and cultured for two further days. On day 9 of the protocol, immature organoids were embedded in Geltrex droplets and subsequently cultured in cerebral organoid differentiation media containing a 1:1 mixture of DMEM/F12 and neurobasal medium supplemented with 0.5 × N2 supplement, 1 × B27 supplement without vitamin A (Thermo Fisher), 0.045 mM 2-mercaptoethanol, 2.5 μg/mL insulin (Roche), Glutamax, and 0.5 × MEM-NEAA in a 10-cm petri dish placed on an orbital shaker in an incubator. Medium was changed every three days. After day 14, cerebral organoid differentiation media were used as described earlier, but they were supplemented with B27 with vitamin A (Thermo Fisher). Forebrain-like organoids were harvested at the indicated timepoints. For cryosectioning, organoids were fixed for 15 minutes with BD Cytofix (BD Biosciences), embedded in Tissue-Tek freezing medium (Sakura), and frozen in isopentane on dry ice. Frozen samples were cut with a cryostat at 12-μm thickness; sections were mounted on superfrost slides and stored at −80°C until staining. In addition, some organoids were fixed in 4% PFA overnight at 4°C and stored in PBS until whole-mount immunofluorescence labeling.
Immunofluorescence labeling and microscopy
For immunofluorescence labeling of fixed cells and tissue slices, samples were thoroughly rinsed with PBS, blocked in PBS, containing 5% normal donkey/goat serum (Jackson Laboratories) and 0.3% Triton X-100 (Carl Roth), and incubated with a mixture of primary antibodies (Table 1) diluted in 0.5% normal serum and 0.03% Triton X-100 at 4°C overnight. After several washing steps with PBS, samples were incubated with corresponding secondary antibodies (Table 1) for one hour at room temperature (RT). Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI, 1:10,000; Sigma-Aldrich) in PBS, and samples were mounted in fluorescence mounting medium (Dako) followed by coverage with cover slips. To control the specificity of immunolabeling, primary antibodies were omitted or tissue from KO mice was stained in parallel. Tissue-specific autofluorescence of human brain tissue was reduced either by using 0.3% Sudan-Black (43) or by capturing images from control channels showing autofluorescence and comparing them with fluorescence channels with the respective immunosignals. Sections were analyzed with an Olympus BX51 epifluorescence microscope, and images were acquired with an XM10 digital camera in combination with CellSens Software (Olympus). For overview images, a Biorevo BZ-9000 microscope (Keyence) was used, while confocal images were acquired by using an Olympus FV1000 laser scanning microscope and Fluoview software (Olympus).
Antibodies
TH, thyroid hormone.
For immunohistochemistry, tissue sections were washed in Tris-buffered saline (TBS; Sigma) and endogenous peroxidases were blocked by incubation with 0.45% hydrogen peroxide (Carl Roth) in methanol for 30 minutes. After rinsing in TBS, immunoglobulin G-blocking was performed with 5% normal goat serum and 0.3% Triton X-100 in TBS. Primary antibodies (Table 1) were diluted in 0.5% normal sera and 0.03% Triton X-100 and incubated overnight, except in negative controls. After rinsing in PBS, slices were incubated with biotinylated secondary antibodies (Table 1) and subsequently with ExtrAvidin-Peroxidases (Sigma-Aldrich), each for one hour at RT. Then, 3,3′-Diaminobenzidin (DAB) substrate (Sigma-Aldrich) was added for visualization. Sections were counterstained with hematoxylin, dried, and embedded in Entellan (Merck KGaA). Results were analyzed and documented with a light microscope (Zeiss Axioplan 2) and ProgRes C3 Capture Pro software.
For whole-mount immunofluorescence labeling, organoids were incubated for at least five hours in PBS-buffered blocking solution containing 1% bovine serum albumin (Sigma), 1% Triton X-100 (Sigma), 4% horse serum (Thermo Fisher), and 0.02% sodium azide (Sigma). Subsequent incubation with primary antibodies in blocking buffer (Triton X-100 concentration reduced to 0.25%) was performed for five days at 4°C with constant rocking. After 8 one-hour washes in PBS and overnight incubation in blocking buffer, organoids were incubated with secondary antibodies in blocking buffer (Triton X-100 concentration reduced to 0.25%) for four days at 4°C with constant rocking. To counterstain cell nuclei, organoids were incubated in DAPI solution (1:5000) for five days at 4°C with constant rocking. After extensive washing in PBS over the course of two days, stained organoids were postfixed in 4% PFA (20 minutes at RT). For tissue clearing, organoids were incubated for three days in refractive index matching solution (0.013 M PBS, containing 88% w/v Histodenz [Sigma], 0.1% Tween 20, and 0.01% sodium azide) and stored in fresh clearing solution at 4°C until imaging.
Semi-quantification and statistical analysis
To compare anti-MCT8 mean fluorescence intensities (MFIs) in between exemplary regions of the murine brain throughout development, images were captured at constant image acquisition settings and exposure times. According to the observed expression pattern of MCT8, analyses comprised the prelimbic cortex, the hippocampal cornu ammonis (CA1-CA4), the Purkinje cell layer of the cerebellum, and blood vessels in P6–P83 wildtype mice (n = 3–4 per group). Sections from KO littermates and WT with omitted primary antibodies served as controls. The MFI was measured with ImageJ (National Institutes of Health, Bethesda, MD) in defined ROIs (Supplementary Fig. S1). In detail, 12 ROIs were measured per animal in consecutive coronal slices with a distance of 48 μm. Statistical differences in between groups were determined with GraphPad Prism 6 (GraphPad Software, Inc., La Jolla) by applying one-way analysis of variance (ANOVA) with Tukey's multiple-comparison test. A p-value <0.05 was considered as statistically significant. Results are presented in scatter plots, with bars visualizing standard deviations of the MFI (mean ± SD).
Results
Validation of MCT8 antibodies
For the current immunofluorescence study, we used a commercially available antibody raised against the intracellular C-terminal epitope of MCT8 (NBP2-57308, Lot no. 100566; Novus). For a first validation of this antibody, we tested its specificity in cell lines and murine brain tissue by comparing it with another well-characterized anti-MCT8 antibody (HPA003353, Lot no. A61491; Atlas) (22,23,44,45) that is no longer available.
MDCK1 cell lines lacking endogenous human MCT8 expression were stably transfected with an expression vector for N-terminal HA-tagged MCT8 or with an empty vector (Fig. 1A). MCT8-expressing cells were stained after cell membrane permeabilization with either anti-MCT8 (NBP2-57308, Lot no. 100566; Novus) or anti-HA antibodies as positive control. Immunofluorescence labeling of cells with anti-MCT8 and anti-HA antibodies showed comparable results, confirming a similar sensitivity to detect the tagged MCT8 protein. Immunolabeling of mock-transfected negative control cell cultures with anti-MCT8 antibodies did not produce a detectable signal (see right panel in Fig. 1A).
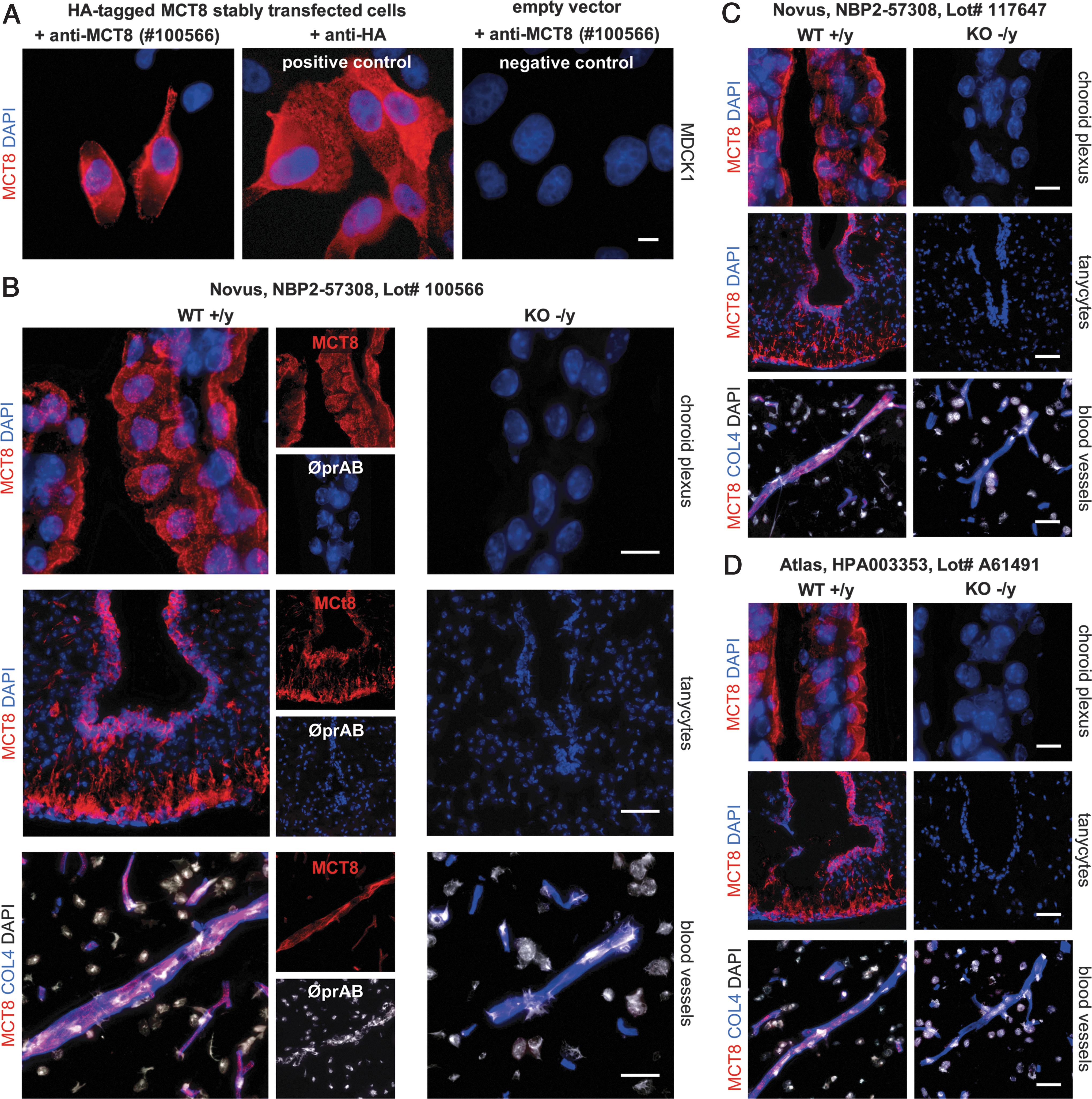
Validation of MCT8 antibodies. (
To further validate the performance of the MCT8 antibody on brain tissue sections, we performed immunolabeling of brain tissue from MCT8-KO mice and WT littermates (P21). For initial validation, we focused on brain structures known to express MCT8, including choroid plexus epithelial cells, tanycytes, and endothelial cells of the BBB (Supplementary Fig. S2). In all cases, we observed a specific signal in WT tissues and a lack of staining in corresponding tissues from KO mice (Fig. 1B–D). Thus, for the subsequent analyses, we used the Novus antibody NBP2-57308 (Lot no. 100566). We also observed a specific signal in human pituitary tissue and confirmed positive MCT8 expression in the anterior pituitary but a lack of MCT8 expression in the posterior lobe (Supplementary Fig. S3).
Optimization of tissue preparation and autofluorescence discrimination
After validating the specificity of the MCT8 antibody, we next performed a series of experiments aiming at an optimization of our labeling protocol with respect to tissue preservation, fixation techniques, and discrimination of autofluorescent signals of murine and human brain tissue samples.
To comparatively assess the influence of different fixation techniques on antigenicity and preservation of morphology, we performed immunolabeling of postmortem human brains by using either fresh-frozen tissue followed by cryosectioning or paraffin-embedded tissue. A qualitative analysis showed remarkably lower MCT8 immunofluorescence intensities for paraffin-embedded tissue with only a minority of vessels displaying positive MCT8 immunolabeling (Supplementary Fig. S4A, B). Therefore, from this point onward, all other analyses of MCT8 expression relied on fresh-frozen tissue.
We next assessed the influence of different fixatives on MCT8 immunofluorescence staining intensities. These experiments were carried out with MCT8-expressing MDCK1 cells. Cell integrity was well preserved after short-term fixation in 100% EtOH (10 seconds) as well as after fixation with 4% PFA (Supplementary Fig. S4B1, B2). For longer-term EtOH fixation (20 minutes), we observed disintegration of cell membranes (Supplementary Fig. S4B1). When comparing the signal intensities after EtOH and PFA treatment, we found that EtOH fixation resulted in much stronger signals compared with PFA (Supplementary Fig. S4B3, B4). Based on these results, a short fixation for 10 seconds in 100% EtOH was selected as a default technique for the subsequent analyses.
To discriminate unspecific autofluorescence signals from specific MCT8 immunolabeling in human postmortem brain sections, we acquired images in additional channels that did not capture signals from any of the secondary antibodies used. We will refer to this approach as control channels. Using this approach on human brain tissue, autofluorescent structures such as lipofuscin deposits and erythrocytes could be distinguished by their exact co-localization in merged-channel images, as indicated by the yellowish appearance, whereas specific immunolabeling still appeared in the color of the respective fluorescence channel (Supplementary Fig. S4C). In addition, we tested whether autofluorescence can be efficiently quenched by treatment with Sudan Black as described elsewhere (30). However, in our hands, protocols involving treatment of the sections with Sudan Black inevitably caused a substantial reduction of the MCT8 immunolabeling intensity (data not shown) and Sudan Black treatment was, therefore, not included in our routine protocols.
MCT8 expression in murine brains
Constant MCT8 protein abundance in barriers of the murine CNS
After proving the specificity of the applied antibodies and optimization of the protocol for immunolabeling, we systematically explored the MCT8 protein expression by immunofluorescence studies throughout the entire murine brain from early developmental ages into adulthood (P6–P83, each group n = 3–4). At all developmental stages analyzed [for species alignments see Pombero et al. (46)], MCT8 was robustly expressed in endothelial cells of the BBB, choroid plexus epithelial cells in all four ventricles (blood-cerebrospinal fluid [CSF] barrier), and tanycytes in the hypothalamic median eminence (blood
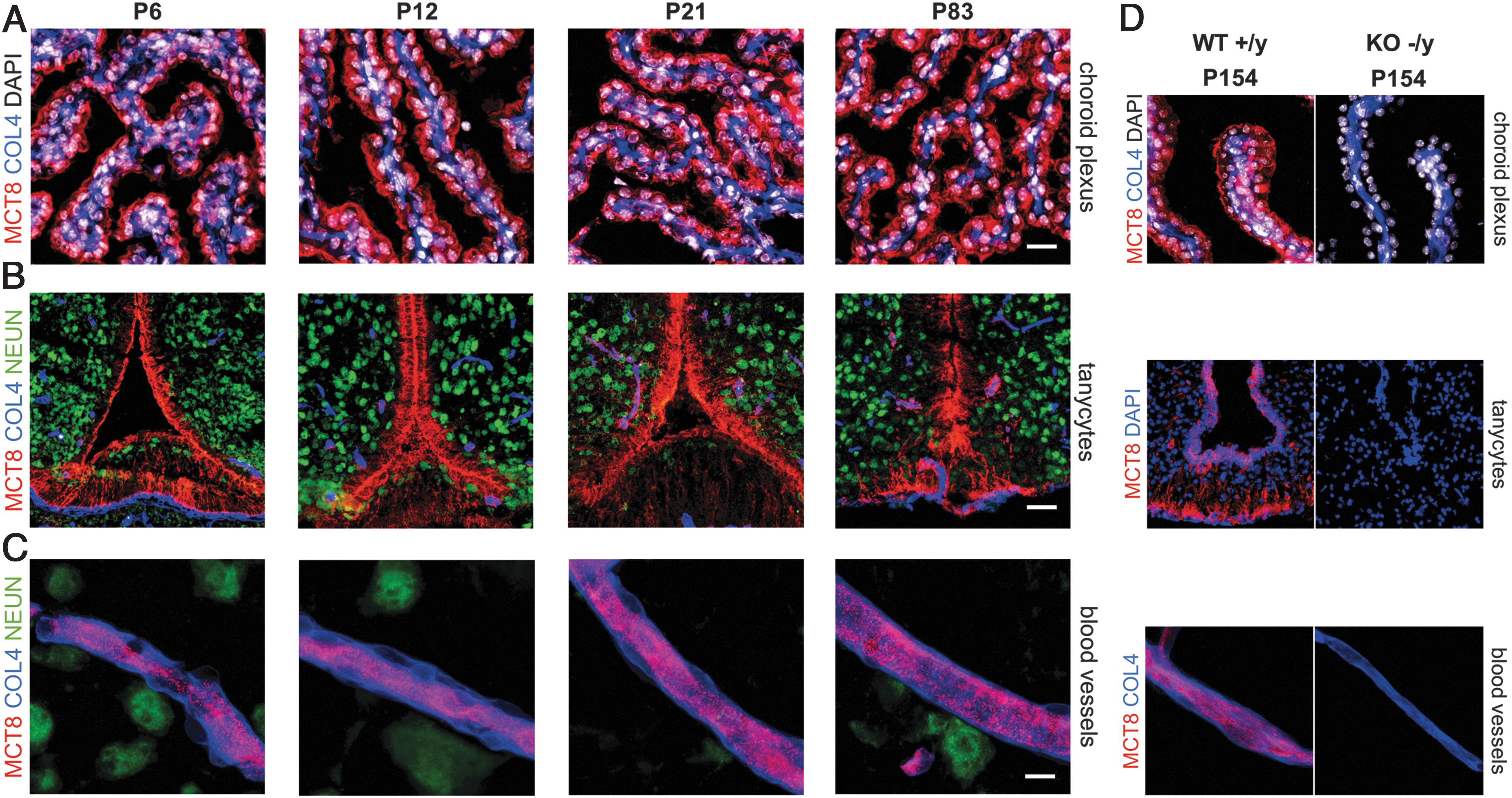
Constant MCT8 protein abundance in barriers of the murine CNS. MCT8 (red) is expressed throughout murine development (P6–P83) in (
In the choroid plexus, MCT8 was observed in apical membranes of choroid plexus epithelial cells (22), facing and producing the majority of CSF (Fig. 2A). As the exposure times were adjusted to the bright immunosignal of MCT8 in choroid plexus epithelial cells, the less intense MCT8 immunoreactivity of the adjacent vessels (anti-COL4) is not captured in the presented images (47). In addition to a membranous cell body labeling, tanycytic processes, projecting from cell bodies lining the third ventricle to blood vessels and hypothalamic neurons (anti-NEUN), also showed strong MCT8 immunosignals (22) (Fig. 2B). MCT8 expression was detected in endothelial cells of all analyzed microvessels throughout the brain (Fig. 2C). Negative control stainings in MCT8-KO mice (P154) and in WT littermates (omission of primary antibodies) verified these results (Fig. 2D). Thus, all barrier structures relevant for brain TH uptake displayed strong and stable MCT8 expression throughout postnatal development.
Developmental decline of murine MCT8 expression in neuronal cell populations
We next extended our systematic immunofluorescence analyses to characterize MCT8 expression in neuronal cell populations. These experiments involved mice from P6 to P83 (n = 3–4 per age group). Brain tissue samples were analyzed in a serial manner from caudal to rostral.
At young stages (P6), MCT8 expression was observed in distinct neuronal cell populations (anti-NEUN) in the following brain areas (Fig. 3 and Table 2): MCT8 was expressed in neuronal cell bodies and projections within the upper layers of the prelimbic cortex (Fig. 3B); in the hippocampus, we observed MCT8 immunosignals in all layers of the four cornu ammonis subfields, comprising neural cell bodies and fibers, but only very little in the hippocampal dentate gyrus (Fig. 3C); we observed MCT8 to be strongly expressed in the cerebellar Purkinje cell layer (Fig. 3D), in neuronal cells of the islands of Calleja (Fig. 3E), of the medial, basomedial, basolateral, and central nuclei and cortical area of the amygdala (Fig. 3F), of the cortical piriform area (Fig. 3G) and olfactory tubercle (data not shown).
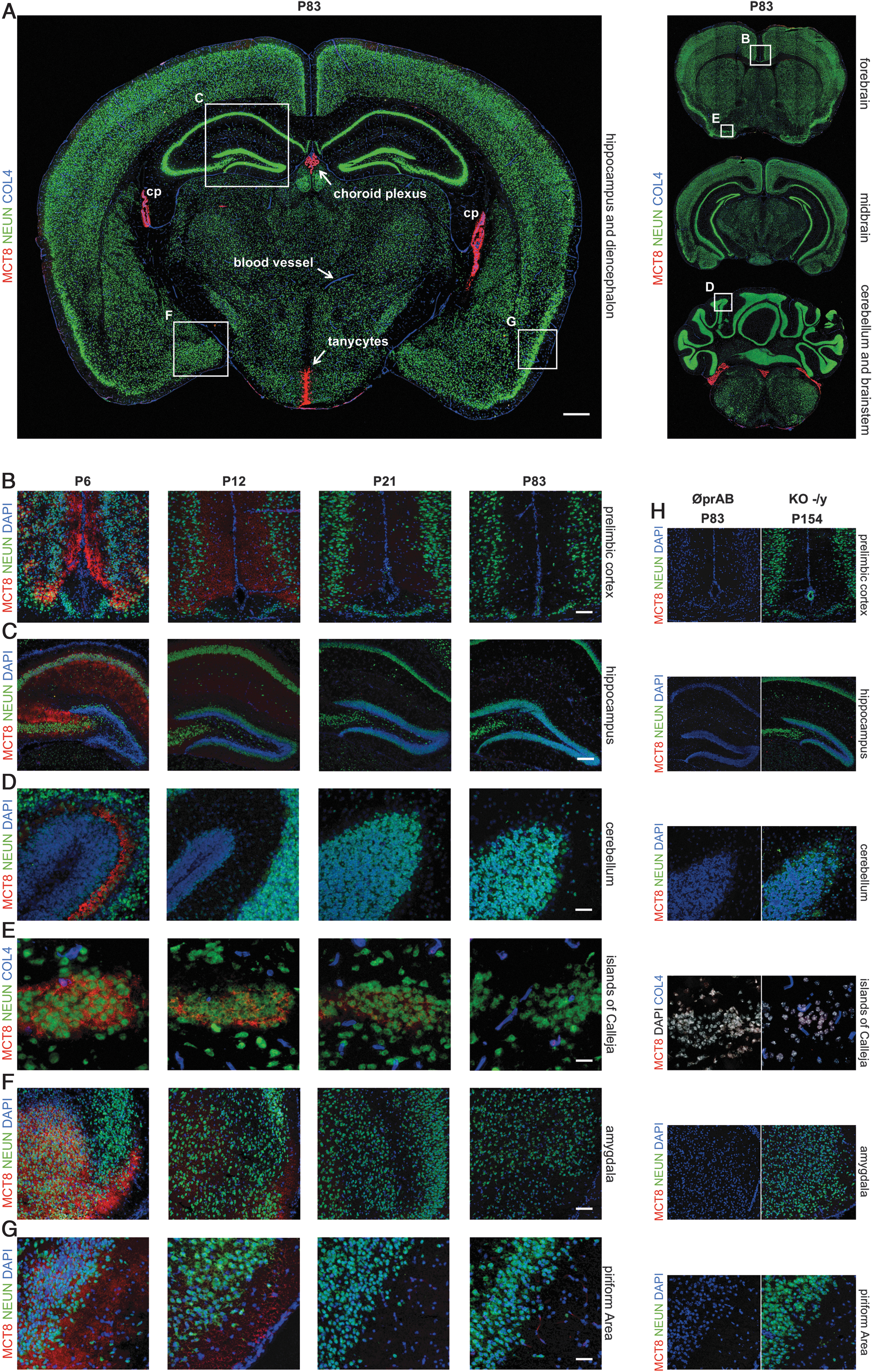
Developmental decline of murine MCT8 expression in distinct neurons. (
Study Synopsis: MCT8 Expression in Murine and Human Cerebral Tissue
We find MCT8 to be constantly expressed in endothelial cells of the BBB in adult mice and humans. Neuronal MCT8 expression is only observed in specific populations during young ages in mice or in neuronal progenitor cells of human cerebral organoids.
+++/++/+/(+)/−, very high/high moderate/low/very low/no MCT8 expression; BBB, blood
However, we also observed a dramatic reduction of neuronal MCT8 expression in all these regions with increasing age. By P83, the intensity of immunolabeling became no longer distinguishable from the background signal detected in MCT8-KO animals.
Consistent endothelial versus decreasing neuronal MCT8 expression with age
Because our qualitative analysis of immunofluorescence of signal intensities indicated marked spatiotemporal changes of MCT8 expression, we next aimed at a quantification of anti-MCT8 MFIs during postnatal murine brain development from P6 to P83 (Fig. 4). For this purpose, the MFI of MCT8 immunosignals was measured in 12 defined ROIs per animal and group (n = 3–4). This quantitative analysis showed that neurons of the prelimbic cortex (Fig. 4A) and the hippocampal cornu ammonis (Fig. 4B) exhibited a rapid and significant decline of MCT8 expression with increasing age. At adult stages (P83), MFIs of neuronal MCT8 immunosignals were found to match background levels of negative controls and KO animals. Purkinje cells in the cerebellum displayed the most rapid reduction of MCT8 expression, with MFI levels measured at P12 approaching those determined for the corresponding ROI in negative control groups (Fig. 4C). In stark contrast to the neuronal cell populations, fairly constant levels of MCT8 protein were observed in endothelial cells of the BBB during development from P6 to P83 (Fig. 4D). For endothelial cells, MFI values did not significantly differ between age groups but were always well above the MFI levels detected in negative controls (n = 3–4, ANOVA followed by Tukey's multiple-comparison test).
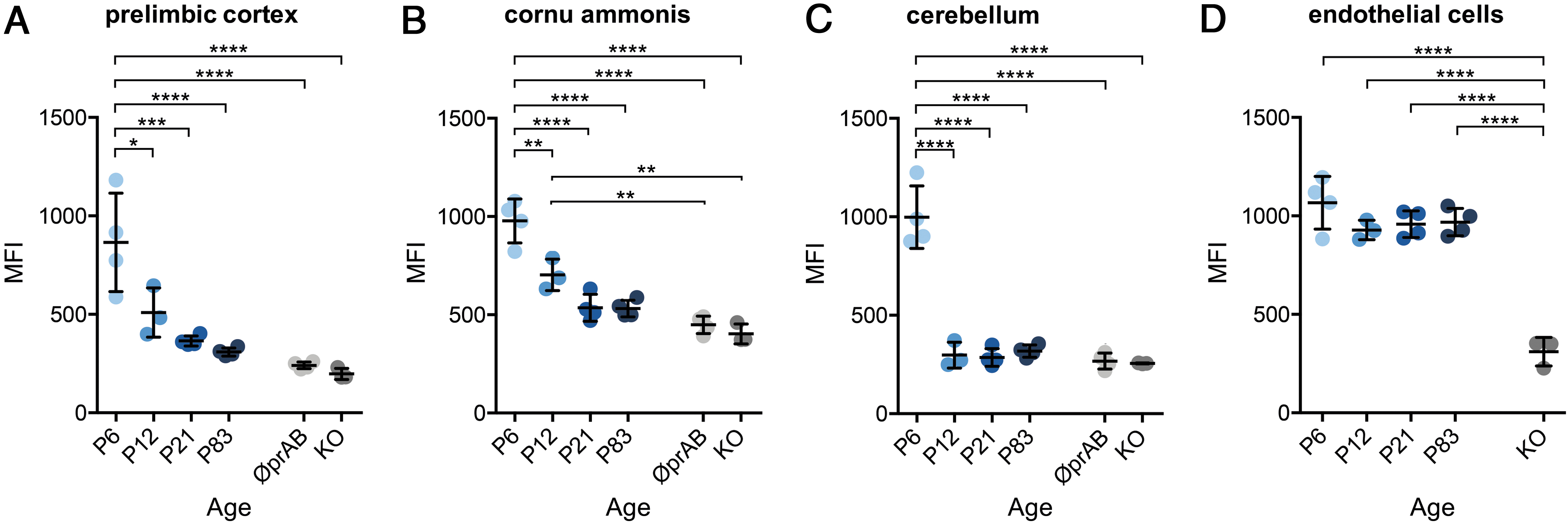
Consistent endothelial versus decreasing neuronal MCT8 expression with age. (
Murine MCT8 is expressed in neuronal projections but not in mitochondria
To further determine the subcellular localization of MCT8 proteins, we additionally labeled the neuronal cytoskeleton (anti-MAP2) to visualize dendritic projections (Fig. 5A, B). We found that MCT8 immunosignals closely mirrored MAP2-immunopositive structures in upper layers of the prelimbic cortex of P6 mice (Fig. 5B). Eventually reaching the cytosol of neurons, TH have to overcome even further barriers such as the porous cell nucleus and mitochondrial membrane to activate DNA-binding TR α and β (6) or mitochondrial TR α variants (48). Confocal laser scanning microscopy revealed that the monocarboxylate transporters MCT2 and MCT4 are concomitantly expressed in plasma as well as in mitochondrial membranes (49). To assess whether MCT8 is expressed in nuclear and mitochondrial membranes as potential sites of T3 action, we assessed MCT8 immunolabeling profiles with respect to markers for mitochondrial membrane channels (anti-VDAC1) and for cell nuclei (DAPI). VDAC1 immunosignals were located around the nucleus, but MCT8 was only detectable in cellular peripheral sites and we observed no colocalization of MCT8 with VDAC1 or DAPI (Fig. 5C, D).
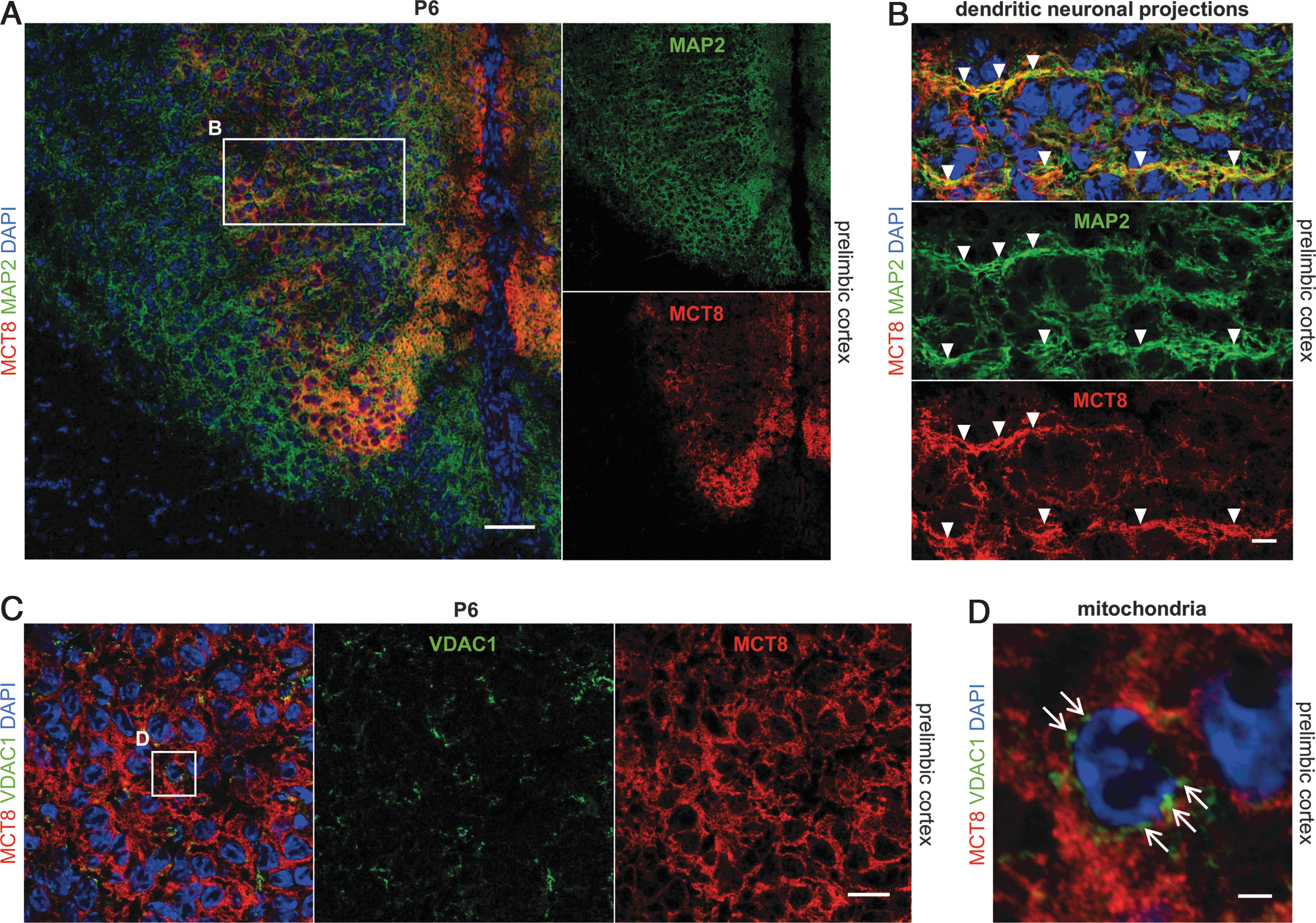
Murine MCT8 is expressed in neuronal projections but not in mitochondria. (
MCT8 expression in human brains
MCT8 protein abundance in endothelial cells of the adult human BBB
To compare the observed murine MCT8 expression patterns with human tissue, we further analyzed 19 brain regions of three elderly human body donors (age 78–82). For this purpose, we applied multiple immunofluorescence labeling of MCT8 in combination with COL4 to identify cerebral vessels. In line with our findings of the adult mouse brain, MCT8 immunoreactivity appeared to be confined to endothelial cells in all adult human brain regions analyzed (Fig. 6A–D). Notably, other components of the NVU such as astrocytes, pericytes, and neurons did not display MCT8 expression (Fig. 6G–I). Unspecific autofluorescence signals were encountered in many brain sections but could be discriminated from specific immunofluorescence labeling on merging with the control channel approach (Fig. 6A–D).
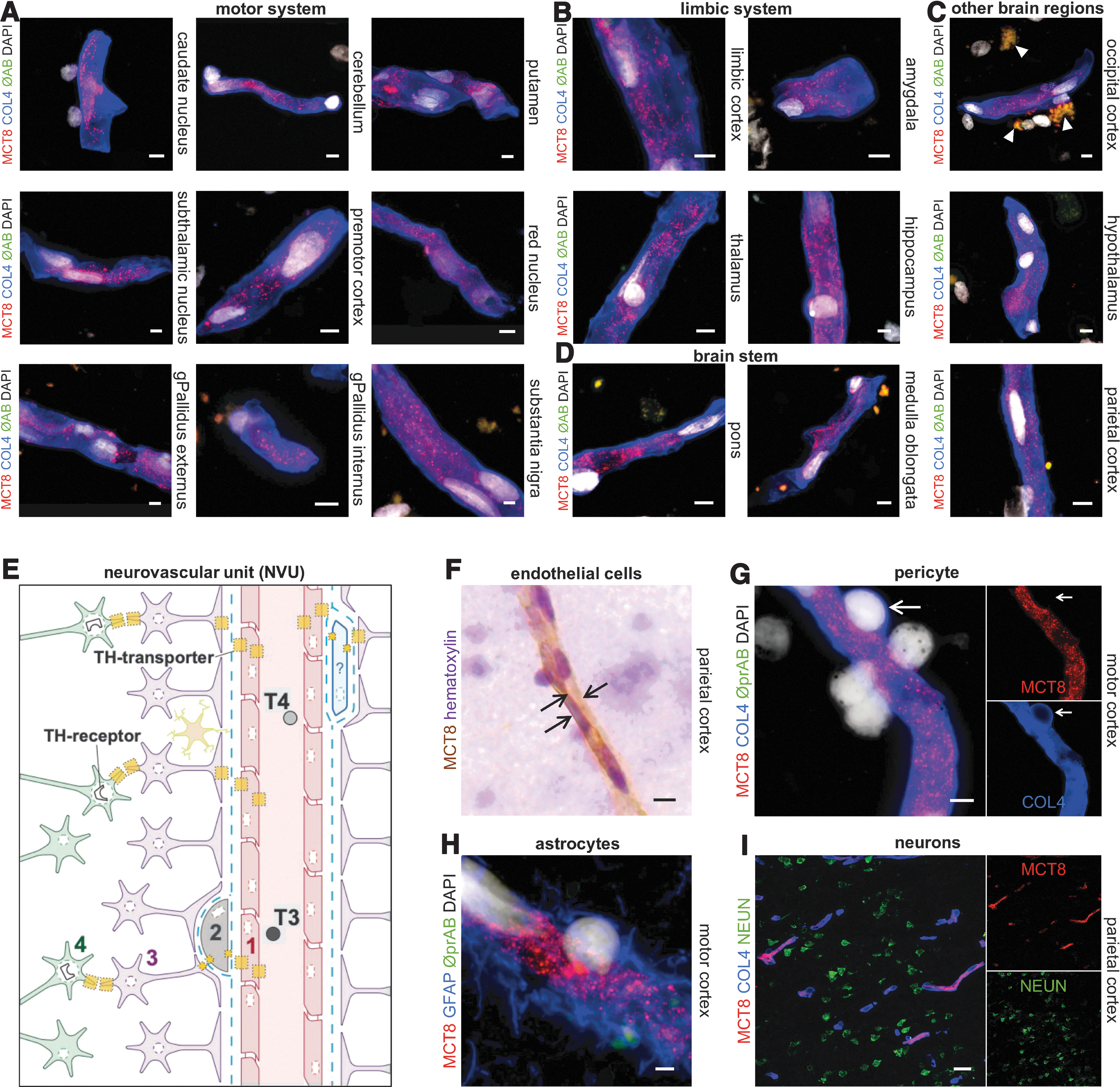
MCT8 Protein abundance in endothelial cells of the adult human BBB. (
Postoperative human brain tissue reveals MCT8 expression only at the BBB
To rule out that the inevitable delay in obtaining human postmortem brain tissue accounts for protein degradation, eventually leading to false negative results on MCT8 expression, we also analyzed snap-frozen neurosurgical brain tissue obtained from a 50-year-old female patient. Analysis of this fresh frozen brain tissue revealed essentially the same MCT8 expression as observed in postmortem brain tissue. Specifically, MCT8 immunosignals were restricted to endothelial cells of the vasculature, whereas neuronal expression was not detectable (Fig. 7A).
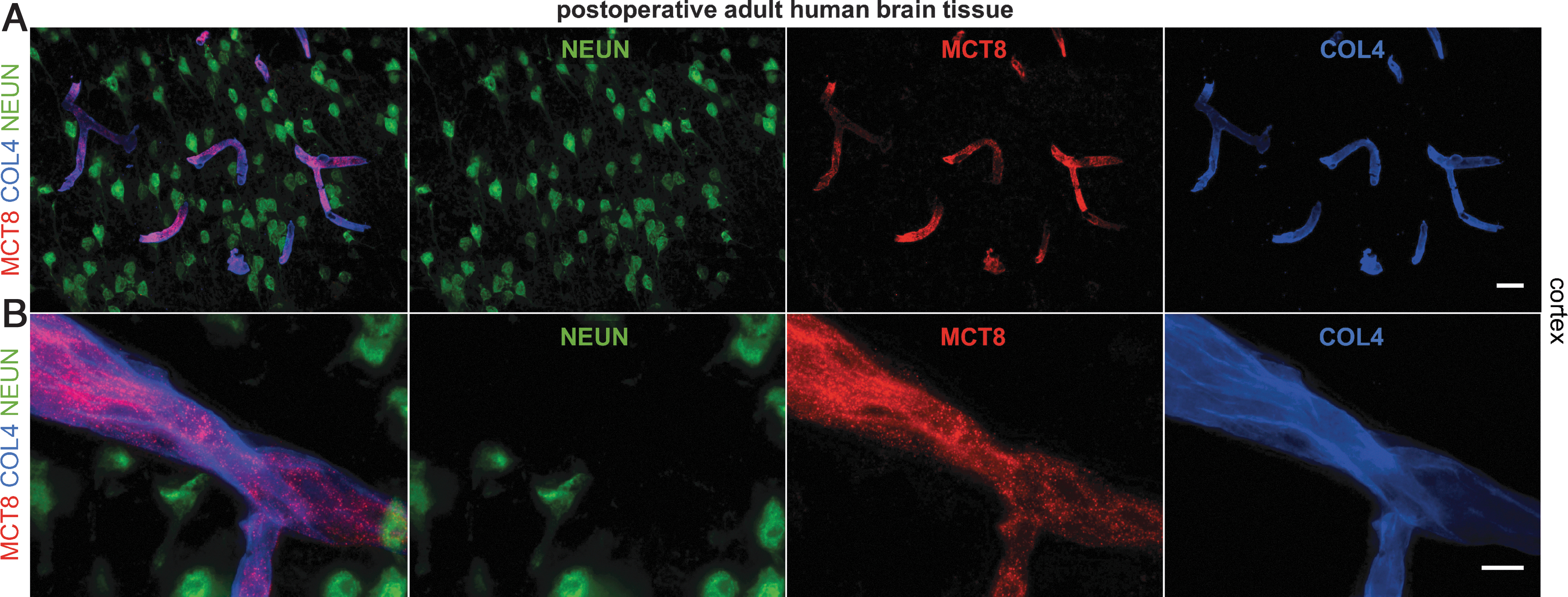
Postoperative human brain tissue reveals MCT8 expression only at the BBB. (
MCT8 is expressed in neuronal progenitor cells of human organoids
Finally, we aimed at elucidating whether human neuronal progenitors during early developmental stages express MCT8 as expected from our murine analysis. Cerebral organoids derived from hiPSC emerged as a new promising in vitro model of embryonic and early fetal brain development. In this study, we used a well-characterized forebrain organoid model (41) that is known to recapitulate cellular diversity and micro-architectural features characteristic of the forebrain, particularly with respect to cortical development.
Forebrain organoids differentiated based on self-organizing principles using limited guiding cues can show a substantial heterogeneity in differentiation rates (41). We, therefore, performed several organoid cultures with two well-characterized hiPSC lines, BIHi004-A and BIHi005-A, and assessed the propensity of these hiPSC lines to differentiate into organoids with a primarily dorsal forebrain (cortical) identity. For this purpose, we analyzed organoids for the expression of general forebrain marker FOXG1 and for PAX6, a marker of dorsal forebrain neuronal progenitors. By focusing our analyses on those organoids, which displayed a grossly normal morphology (round shape, smooth surface, neuroepithelial buds projecting out of the surface), we found that both hiPSC lines generated organoids with the expected dorsal forebrain patterning (Supplementary Fig. S5). Specifically, we observed that the majority of organoid tissues displayed strong FOXG1 expression in both progenitor and early neuronal cells (Supplementary Fig. S5E–G) and that SOX2+ progenitors robustly co-expressed the dorsal forebrain marker PAX6 (Supplementary Fig. S5A, B). However, we also noticed that organoids frequently contained additional tissue types that did not express any of the dorsal forebrain markers analyzed (PAX6, FOXG1) or lacked expression of neuronal differentiation markers (SOX2, PAX6, doublecortin [DCX], TUJ1) (Supplementary Fig. S6L–N and data not shown). The molecular identities of these additional tissues are not yet known. Regions of unknown identity accounted in most cases for a small proportion of organoid tissue and were mainly found in the periphery of the main organoid body.
We next performed a series of immunolabeling studies to verify that organoids cultured under these experimental conditions generate layered structures resembling the in vivo cortical cytoarchitecture. In accordance with previous descriptions of cerebral organoid cytodifferentiation (41), we confirmed that the large majority of SOX2+/PAX6+/Nestin+ neuroepithelial progenitors were organized in multiple, circular, rosette-like substructures by three weeks of culture (Supplementary Figs. S5A′, B′ and S6A). Co-staining of SOX2 and Nestin revealed that these rosette-like substructures show the hallmarks of pseudo-stratified ventricular zone-like regions with dense vertical columns of SOX2+/Nestin+ progenitor cells (Supplementary Fig. S6A). When analyzing these early organoids for expression of DCX, a marker of early immature cortical neurons, we found that neurogenesis was minimal at three weeks of culture (Supplementary Fig. S6B). If present, DCX expression was limited to regions close to the surface in all three-week-old organoids analyzed.
However, as early as four weeks of culture, organoids displayed the generation of well-defined multi-layered cortical-like structures characterized by an enlarged layer of DCX+ neurons surrounding SOX2+ ventricular zone-like regions (Supplementary Fig. S6H–K). The emergence of cortical-like structures displaying features of in vivo germinal (ventricular, subventricular layers) and cortical plate zones was further verified by whole-mount immunolabeling of organoids with additional markers of neuronal progenitors (SOX1) and neurons (TUJ1) (Fig. 8A). We noticed that the majority of organoids preselected based on the aforementioned gross morphological characteristics showed these features of distinct progenitor zones surrounded by neuronal populations throughout the entire organoid tissue (Fig. 8A).
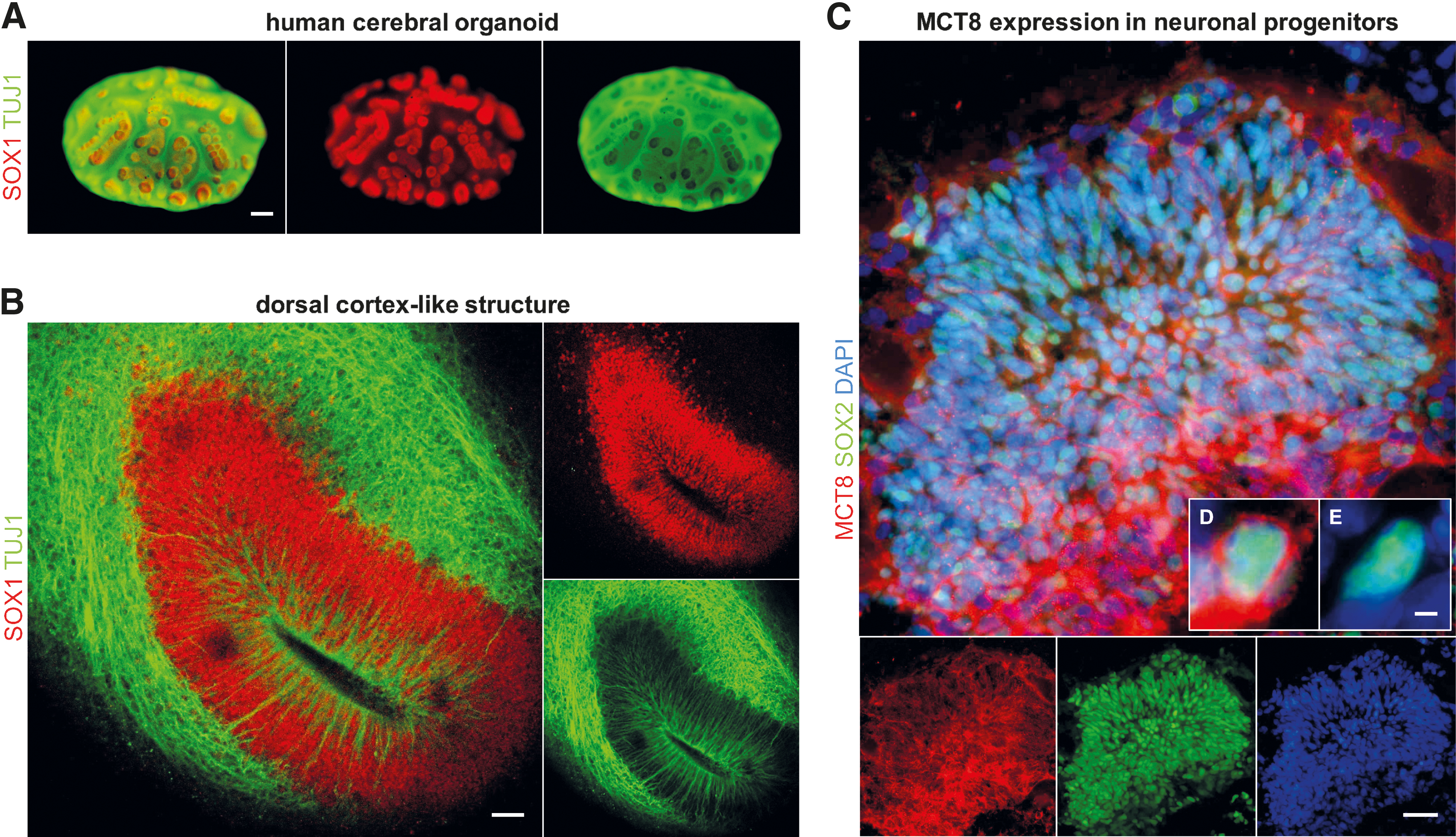
MCT8 is expressed in early neuronal progenitor cells of human forebrain-like organoids. (
By culture day 50 (corresponding to ∼13/14th fetal postconceptional week) (50), formation of larger radially organized forebrain-like tissue (greater than 400 μm in diameter) was evident and it showed hallmarks of dorsal cortical development, including a fluid-filled luminal compartment, an apically located layer of progenitors reminiscent of the ventricular zone, and a neuron-populated basal region (Fig. 8B).
To probe the general utility of cerebral organoid models for studies on TH transporter expression, we applied our MCT8 immunofluorescence protocol to tissue slices of early organoid stages (culture day 30, approximately ninth fetal postconceptional week) (51). Specifically, we assessed whether neuronal progenitors forming the pseudo-stratified ventricular zone-like structures express MCT8 as anticipated from previous studies in primary fetal tissues (22,24 –26,52,53). Immunolabeling of MCT8 and SOX2 in tissue sections of 30-day-old organoids showed broad expression domains of MCT8 overlapping SOX2+ rosette-like structures (Fig. 8C). However, on closer inspection, we noticed that not all SOX2+ progenitors showed positive MCT8 staining, demonstrating that the rosette-like structures are composed of mixed populations of MCT8+/SOX2+ and MCT8−/SOX2+ cells (Fig. 8D, E). The positive detection of MCT8 expression in cortical progenitor cells was highly reproducible, as it was observed in six out of six organoids analyzed, with organoids derived from three independent cultures.
Discussion
Impairment of TH transport from the systemic circulation into brain tissue leads to neurodevelopmental disorders as evident in patients with MCT8 deficiency affected by severe intellectual disability, truncal hypotonia, limb dystonia, and spasticity. The pathophysiology of the major symptoms of these children is still poorly understood and, therefore, strategies to develop effective therapies are currently unavailable. To define the cellular, regional, and temporal cerebral MCT8 protein expression, we performed a comprehensive interspecies immunofluorescence study with specific anti-MCT8 antibodies (Table 2). In serial sections of murine brains, we found MCT8 to be expressed at highest levels in critical barriers for TH uptake, including the choroid plexus epithelial cells, tanycytes, as well as in endothelial cells of the BBB (Figs. 2 and 4). For the MCT8 abundance in endothelial cells of the BBB, we observed uniform high expression in all brain regions analyzed and no changes during postnatal development in mice. A different situation was evident for MCT8 expression in neuronal cell populations, as we observed a dramatic decline of immunosignals for MCT8 in all analyzed neuronal populations until P83, in contrast to the very stable immunosignal, was detectable in endothelial cells across all ages (Figs. 3 and 4). Our analyses of adult human samples showed a very similar MCT8 expression profile characterized by robust MCT8 expression in endothelial cells of the BBB but a lack of immuno-detectable MCT8 in neuronal populations (Figs. 6 and 7). Conversely, in cerebral organoids as a model of early human neurodevelopment, we observed strong MCT8 expression in early neuronal progenitor populations (Fig. 8). Collectively, these data support a view that there are profound differences in the developmental expression dynamics between neuronal cell populations as the final target of TH action and the various cell types that function as barriers of TH uptake by the brain. This observation was clearly corroborated for murine brain tissue, and our data indicate that a similar scenario might hold true for human brain tissue.
MCT8 protein expression in the brain
Information about the spatiotemporal expression of MCT8 protein is still sparse and scattered among individual studies. In Supplementary Tables S1 and S2, we summarize the main findings of our and of previously published studies on MCT8 protein expression. One major consensus among these studies is that MCT8 is strongly expressed in structures with barrier function, including endothelial cells of the BBB (22 –26,47,54,55). Study results, however, differ with respect to the detectability of MCT8 in the neuronal population, particularly at late postnatal stages. In our study, immunolabeling of neuronal cell populations in adult brain tissues yielded mostly negative results while some studies observed detectable levels of MCT8 protein in specific brain regions (21 –25,27). For example, in one of the most comprehensive analyses, Wirth et al. reported MCT8 protein expression in adult murine cerebral cortex, hippocampus, dentate gyrus, and cerebellar cortex based on IHC staining (22). One likely explanation of the discrepancy to our current analyses is that enzymatic amplification of signals during the IHC procedure allows to detect even very low levels of MCT8 protein. Despite these differences, we believe that analyzing discrete brain regions across developmental stages presents a specific strength of our study and strongly corroborates a dramatic decrease in neuronal MCT8 expression levels from young to adult stages. At this point, it remains elusive whether MCT8 expression at the detection limit restricts nuclear TH availability. Van Mullem et al. described overexpression of MCT8 to increase TH influx and efflux in vitro (56); in that experiment, intracellular T3 concentrations remained stable. One might speculate that higher expression of MCT8 in early developing neurons may only facilitate a faster dynamic adaptation of acute T3 need, rather than causing increased intracellular T3 concentrations. In that case, reduced MCT8 expression would result in lower T3 uptake but may not affect the overall intracellular T3 concentration.
In consideration of comparable expression patterns of MCT8 in mice and humans, discrepancies in neurological phenotypes of MCT8-KO mice without obvious motor impairments and MCT8-deficient patients, who reach very early steps of motor development only, cannot be fully explained based on the currently reported MCT8 expression patterns alone (14). Other TH transporters such as Lat2 in neurons (22) or Oatp1C1 in the BBB (54) are candidates for compensating rodent Mct8 deficiency. The additional deletion of Oatp1C1 in a double KO mouse model completely restricted the uptake of radiolabeled TH into brain homogenates (29). Whether this model corresponds to the suspected regionally differently impaired TH uptake in MCT8-deficient patients remains unclear, since the knowledge about spatiotemporal expression patterns of the variety of TH transporters in both species is still limited. For a more comprehensive understanding, additional finely grained expression analyses at early stages are needed for other transporters.
Cues from MCT8 expression profiles for the pathophysiology of MCT8 deficiency
As noted earlier, our study indicates marked differences in the developmental expression dynamics of MCT8 between barrier structures and neuronal cell populations. Such differential expression profiles may have several implications on the pathophysiology of MCT8 deficiency. First, the ubiquitous endothelial MCT8 expression does not completely explain the clinical phenotypic spectrum of patients with MCT8 deficiency (23,54). If MCT8 deficiency led to a strictly limited TH transport through the BBB, the neurological phenotype of MCT8-deficient patients would be expected to resemble symptoms of a globally hypothyroid brain, including fatigue and depression, both of which are not described/observed in patients with MCT8 deficiency [own unpublished clinical experience (12)]. Although an accurate mapping of neurological symptoms is very difficult due to very low developmental ages of these patients and the number of observations is low (12,57,58), at present, the increase in patients with an established diagnosis and the growing cohorts of MCT8-deficient patients may provide an opportunity to further refine the characterization of the neurological phenotype. In this context, regionally different deficits in TH supply need to be considered as an important factor contributing to the pathophysiology and phenotype of MCT8 deficiency.
The strong expression of MCT8 in the choroid plexus raises the question about its role in TH transport to the CSF. Uptake studies in rats and chicken revealed faster and higher accumulation of intravenously injected radiolabeled TH in the choroid plexus in comparison to other brain regions (59 –61). Although highly speculative, this could be a result of TH leakage through characteristic convolutions of blood vessels in this region and may point toward TH secretion to the liquor. Due to the high co-expression of type 3 iodothyronine deiodinase in choroid plexus ependymal cells (53), the actual passage of TH to the CSF is questionable; however, lowered T4 liquor concentrations have, indeed, been reported in one patient with MCT8 deficiency (62). Hence, this TH route into the brain may also be considered in the pathophysiology of MCT8-deficient patients.
Although strong MCT8 protein expression is detectable in human tanycytes (21,27), it is unclear how MCT8 deficiency would restrict TH transport from blood to hypothalamic neurons. The TH supply of neurons in the paraventricular nucleus (PVN) negatively regulates their adenohypophyseal secretion of thyreotropin-releasing hormone, stimulating the release of thyrotropin (TSH) by the anterior pituitary and subsequently TH by the thyroid (63). If the tanycytic TH transport to the PVN was critically limited in MCT8 deficiency, the lack of negative feedback led to higher systemic levels of TSH that are not monitored in patients. Thus, alternative tanycytic TH transports are likely to exist.
MCT8 expression in human cerebral organoids
The highly dynamic age-dependent MCT8 protein levels in neurons emphasize the need for detailed studies on early stages of human neuronal development. After completion of our analyses, a very recent study by López-Espíndola et al. addressed this point by analyzing human fetal brain tissues from gestational week 14 to 38 for expression of MCT8 and OATP1C1 (52). This work showed MCT8 expression in barrier structures, including choroid plexus epithelial cells, tanycytes, and endothelial cells of the BBB at all stages analyzed. In addition, MCT8 was also localized in specific neuronal populations as well as in radial glial cells. The latter observation is of special interest, as it reinforces the view that MCT8-mediated TH transport is relevant for very early stages of neuronal development (52).
Given the restricted availability of primary human fetal tissue, cerebral organoids derived from hiPSC have recently been proven to provide unprecedented opportunities to study various aspects of embryonic and early fetal brain development in a human context (64). As a starting point for a more comprehensive analysis of MCT8 expression in specific subpopulations during forebrain-like organoid development, we show in our study that MCT8 is already expressed in subpopulations of neuronal progenitor cells at early stages of organoid differentiation (Fig. 8). This finding corresponds well with the detection of MCT8 in radial glial cells (52) and is in line with other studies of primary fetal human brain detecting MCT8 in distinct CNS precursor cells (22,24 –26). Although cerebral organoid cultures currently still lack a concomitant development of a vascular network, they are proven models to study various neurodevelopmental defects (65) and might present a valuable approach to decipher early neuronal development under conditions of MCT8 deficiency.
Implications for therapy strategies of MCT8 deficiency
For the development of novel treatment strategies, the cellular MCT8 localization is of utmost importance. It needs to be considered as to which TH transport step from the blood to neurons over all cell types of the NVU is impaired in MCT8 deficiency and, therefore, has to be overcome by a therapeutic strategy. Our data suggest that in MCT8 deficiency, TH transport could be mainly defective over two barriers if not compensated by other transporters: (i) at the level of endothelial cells at the BBB and (ii) at the level of neuronal plasma membranes within distinct brain regions during early developmental stages. Given that our human analyses were limited to adult brain tissue only, further knowledge about transient changes of human neuronal MCT8 expression perinatally (53) will be pivotal for identifying critical periods for therapeutic interventions pre- and postnatally.
However, the intracellular TH transport over subcellular membranes to DNA-bound TH receptors may be enabled primarily by other TH transporters or nuclear pore complexes, as we observed MCT8 immunosignals neither in mitochondrial nor in nuclear localizations (Fig. 6).
Our observations of profoundly different developmental expression dynamics of MCT8 in barrier structures versus neuronal cell populations are deemed important for a better understanding of the specific neurodevelopmental phenotypes in patients with MCT8 deficiency. Our results highlight the need for more systematic studies, particularly on the very early expression profiles of MCT8 and other TH transporters in different model systems to identify sensitive neuronal cell populations and critical time windows for treatment strategies.
Footnotes
Acknowledgments
The authors thank A. Ehrlich, M. Oehme (University of Leipzig), and S. Jyrch (Charité – Universitätsmedizin Berlin) for excellent technical assistance; D. Zwanziger (University of Essen) and G. Krause (FMP Berlin) for providing antibodies; and the team of the BIH Core Facility Stem Cells funded by the BIH for support of the induced pluripotent stem cells-related work.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was partially supported by the Berlin Institute of Health [BIH Medical Student Research Stipends to N.-M.W., BIH-Charité Clinical Scientist Program to P.M.], by the Deutsche Forschungsgemeinschaft [DFG, ThyroidTransAct SPP1629 BI893/5-2 to H.B., KR1710/5-1 to H.K.], [DFG, KU 2673/2-2 to P.K.], [DFG, SFB 1052 to I.B.], and by the Bundesministerium für Bildung und Forschung [BMBF, 16GW0191 to P.M. and H.S.].
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2