
Abstract
Background:
MicroRNAs (miRNAs) are a class of critical epigenetic regulators involved in several autoimmune diseases. Our previous study reported an miR-326-induced increase in T helper (Th) 17 cells in a mouse model of Hashimoto's thyroiditis (HT), but the pathogenic effect of miR-326 in HT patients has not been verified. The goal of the present study was to explore the pathogenic role of miR-326 and its underlying molecular mechanism in HT patients.
Methods:
A total of 58 HT patients and 55 normal controls were enrolled in this study. We examined whether Th17 cells and miR-326 were aberrantly altered in the peripheral blood mononuclear cells (PBMCs) of HT patients with flow cytometry and real-time polymerase chain reaction. Levels of membrane interleukin (IL)-23R (mIL-23R) were determined by flow cytometry and Western blot to explore the critical role of mIL-23R in the development of Th17 cells. Isolated CD3+ T cells were used to further investigate the ectodomain shedding of mIL-23R by a disintegrin and metalloprotease (ADAM17). Furthermore, miR-326 inhibitor and mimics were transfected into PBMCs derived from HT patients and healthy controls to verify the regulation of ADAM17 by miR-326.
Results:
We observed elevated miR-326 levels in the PBMCs of HT patients compared with those in the PBMCs of healthy controls. Consistent with IL-23-induced STAT3 overactivation, substantially more HT patient-derived PBMCs differentiated into Th17 cells under polarization culture conditions, which may, at least in part, have resulted from enhanced mIL-23R levels. Furthermore, ADAM17, an ectodomain sheddase of mIL-23R, was targeted and negatively regulated by miR-326. Inhibiting ADAM17 might attenuate the ectodomain shedding of mIL-23R.
Conclusions:
Our findings suggest that the effect of miR-326 on the IL-23/IL-23R/Th17 cell axis in HT patients might be partially due to the targeting of ADAM17.
Introduction
Hashimoto'
Recently, the differentiation of T cell subsets by epigenetic modifications, including those made by microRNAs (miRNAs), has attracted attention. As a class of endogenous single-chain, noncoding RNAs ∼22 nucleotides long, miRNAs bind the 3′-untranslated regions of their target mRNAs to prevent their translation or cause their degradation in a series of physiological and pathological processes (6). MiR-326, one of many miRNAs, was shown to contribute to the expansion of pathogenic Th17 cells in multiple sclerosis and mouse models (7). Similar results were also found in other autoimmune diseases, such as Behcet's disease (BD) (8). Our previous study in NOD.H-2h4 mice revealed that the differentiation of Th17 cells in iodine-induced thyroiditis can be promoted by miR-326 (9). However, little research in HT patients has been conducted so far.
The expansion and survival of Th17 cells rely on their responses to the cytokine IL-23 (10). As a member of the IL-12 family, IL-23 consists of a unique p19 subunit and the p40 subunit of IL-12. Activation of STAT3 is a key event in Th17 lineage specification (11). By engaging with its receptor, membrane IL-23R (mIL-23R), IL-23 promotes the phosphorylation of STAT3 (pSTAT3) and subsequently induces the differentiation, maturation, and expansion of Th17 cells (12,13). Recently, Figueroa-Vega et al. (14) detected an increased expression of IL-17 along with elevated numbers of IL-23R(+) cells in thyroid tissue of HT patients. A neutralizing antibody targeting the IL-23/IL-23R/Th17 cell signaling axis has been shown effective in treating several autoimmune diseases (15,16). IL-23R-deficient cells have been reported to be resistant to Th17-mediated autoimmune diseases such as systemic lupus erythematosus (SLE) (17), which may demonstrate a relationship between IL-23R and autoimmune diseases. All these studies suggest a critical role of IL-23R in the IL-23/IL-23R/Th17 cell axis.
A recent study reported that surface IL-23R is a substrate for a disintegrin and metalloprotease, ADAM17 (18), a membrane-anchored enzyme that sheds the ectodomains of nearby membrane-bound proteins into soluble proteins (19). Currently, >20 substrates, including cytokines, cytokine receptors, cell adhesion molecules, and chemotactic factor receptors, all of which are critical to immune responses, are known to be processed by ADAM17 (20). Interestingly, ADAM17 was identified as a target of miR-326 by bioinformatic analysis, and the regulation of ADAM17 by miR-326 has been reported in lung adenocarcinoma (21). These findings inspired us to hypothesize that miR-326 may regulate the IL-23/IL-23R/Th17 cell axis by targeting ADAM17 to play a critical pathogenic role in the development of HT.
Herein, we explored the expression profile of miR-326 in HT patients. In addition, peripheral blood mononuclear cells (PBMCs) derived from HT patients and healthy volunteers were used to explore the potential molecular mechanism by which miR-326 targets ADAM17 to promote the expansion of Th17 cells in HT patients in vitro.
Materials and Methods
Subjects and samples
Fifty-eight newly diagnosed adult HT patients (55 females and 3 males) were enrolled in this study. The HT diagnosis criteria were as follows: diffuse goiter, increased levels of serum antithyroglobulin antibody (TgAb) and/or thyroperoxidase antibody (TPOAb), and heterogeneous echotexture in those who accepted thyroid ultrasound examination. We also recruited 55 age-, sex-, and body mass index (BMI)-matched healthy adult volunteers (51 females and 4 males) as normal controls (NC). Participants with other autoimmune diseases (such as SLE), chronic organic diseases, tumors, and infectious diseases were excluded from the present study. None of them was taking drugs that affect thyroid function or the immune system. This study was approved by the Ethics Institutional Review Board of China Medical University before recruitment. All the participants signed informed consent before enrollment.
Elbow venous blood samples were collected at 8 AM after overnight fasting. Serum and plasma were collected and stored at −80°C until use. PBMCs were separated from fresh anticoagulated blood by Ficoll density gradient centrifugation for further study.
Laboratory measurements
The electrochemiluminescence immunoassay (Roche Diagnostics, Switzerland) was performed as described previously (22) to measure serum levels of free triiodothyronine (fT3), free thyroxine (fT4), thyrotropin (TSH), TPOAb, and TgAb.
Isolation of total RNA and quantitative real-time polymerase chain reaction
Total RNA, including miRNA, was extracted from PBMCs with the TRIzol reagent (Invitrogen). A small RNA reverse transcriptase kit (Tiangen, China) was used for miRNA reverse transcription. Then, real-time polymerase chain reaction (RT-PCR) was performed with an miRcute miRNA qPCR detection kit, a random primer, and a unique miR-326 primer (Tiangen) according to the manufacturer's protocol. mRNA was reverse transcribed with a PrimeScript RT reagent kit (TaKaRa, Japan), and PCR amplification was carried out using a SYBR® Premix Ex TaqTM II kit (TaKaRa). U6 small nuclear RNA and GAPDH were used as the internal controls for miRNA and mRNA, respectively. Primer sequences are listed in Table 1. These reactions were conducted according to the manufacturer's instructions with a LightCycler 480 RT-PCR system.
Primer Sequences for Quantitative Real-Time Polymerase Chain Reaction
Enzyme-linked immunosorbent assay for cytokines
Serum interferon (IFN)-γ levels were detected with a human IFN-γ enzyme-linked immunosorbent assay (ELISA) kit (BCBIO Biotechnology, China). Both the intra-assay and interassay coefficient of variation (CV) values were <15%. Levels of IL-17A and soluble IL-23R (sIL-23R) in the plasma and culture supernatants were analyzed with commercial ELISA kits (R&D Systems). Both the intra-assay and interassay CV values were <10%. All tests were performed according to the manufacturer's instructions.
In vitro Th17 polarization
PBMCs isolated from fresh blood were cultured to induce Th17 polarization. Briefly, cells were incubated at 37°C in 5% CO2 for 72 hours with plate-bound anti-CD3 antibody (10 μg/mL; BioLegend) and soluble anti-CD28 antibody (1 μg/mL; BioLegend) in the presence of recombinant human IL-6 (20 ng/mL; R&D Systems), IL-23 (10 ng/mL; R&D Systems), TGFβ (10 ng/mL; R&D Systems), neutralizing anti-human IFN-γ (1 μg/mL; R&D Systems), and neutralizing anti-human IL-4 (1 μg/mL; R&D Systems). After treatment, the cells were collected for further analysis.
Flow cytometry analysis
For intracellular cytokine assays, cells were incubated with a cell activation cocktail (with BFA) at 37°C in 5% CO2 for 5 hours. Then, the cells were incubated with an FITC-conjugated anti-human CD4 antibody (BioLegend) for 20 minutes at room temperature in the dark. Cells were then fixed and permeabilized with a Fix & Perm cell permeabilization kit (Invitrogen). The cells were subsequently stained with PE-conjugated anti-human IL-17A or PE-conjugated anti-human IFN-γ (both from BioLegend). For Treg assays, cells were incubated with an FITC-conjugated anti-human CD4 antibody and an APC-conjugated anti-human CD25 antibody (BioLegend). Fixation and permeabilization were performed with FoxP3 fixation/permeabilization buffer (BioLegend). Cells were then stained with PE-conjugated anti-human Foxp3 antibody (BioLegend).
For STAT3 phosphorylation assays, frozen PBMCs were thawed and incubated in X-VIVO™ 15 medium for one hour as described previously (23). Then, the cells were or were not stimulated with IL-23 (R&D Systems) overnight. After fixation, cells were permeabilized with True-Phos™ Perm Buffer (BioLegend) according to the manufacturer's protocol. Then, cells were stained with a PerCp/Cy5.5-conjugated anti-human CD4 antibody, an FITC-conjugated anti-human CD8 antibody, and a PE-conjugated anti-STAT3 Phospho (Tyr705) antibody (all from BioLegend). To analyze surface proteins, the cells were stained with a PE-conjugated anti-human ADAM17 antibody or a PE-conjugated anti-human IL-23R antibody (both from R&D Systems). Data acquisition was performed with a BD FACSCalibur flow cytometer, and FlowJo 7.6 software was used to analyze the data.
Western blot analysis
PBMCs were lysed, the total protein was extracted with a total protein extraction kit (KeyGen Biotech, China), and the concentration was determined with a bicinchoninic acid assay (KeyGen Biotech). After electrophoresis in 10% SDS-PAGE, the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad). The membranes were washed with tris-buffered saline-Tween and then blocked with 5% dried skim milk or bovine serum albumin. Incubation with specific primary antibodies against STAT3 (1:1000; Abcam, United Kingdom), pSTAT3 Tyr705 (1:800; Cell Signaling Technology), ADAM17 (1:1000; Santa Cruz), or GAPDH (1:1000; Santa Cruz) was followed by incubation with a horseradish peroxidase-conjugated secondary antibody (1:5000; Bioss, China). Signals were detected with a chemiluminescence detection system (Santa Cruz), and images were analyzed with ImageJ software.
CD3+ T cell isolation and ADAM17 suppression assay
Isolation of human CD3+ T cells from fresh PBMCs was performed with a MojoSort™ Human CD3 T Cell Isolation Kit (BioLegend) according to the manufacturer's instructions. Pure CD3+ T cells were detected by flow cytometry. An ADAM17 suppression assay was conducted as described previously (23). Isolated CD3+ T cells were cultured for 30 minutes in 200 μL X-VIVO 15 medium containing 20% fetal bovine serum at a density of 107/mL in the presence or absence of TNF-α protease inhibitor 1 (TAPI-1, 20 mM; Selleck), an ADAM17 inhibitor. Cells were then incubated with a plate-bound anti-CD3 antibody (10 μg/mL) and a soluble anti-CD28 antibody (1 μg/mL) for 4 hours. The treated cells were collected for further flow cytometry.
Transfection of miR-326 mimics or inhibitor
Fresh PBMCs were transfected with miR-326 mimics or an inhibitor to overexpress or inhibit the expression of miR-326, respectively. The miR-326 inhibitor and mimics, as well as the respective controls, were synthetized by GenePharma (Suzhou, China), and the sequences are listed in Table 2. Transfection was conducted according to the manufacturer's protocol. Briefly, miR-326 mimics (20 nM), mimic controls (20 nM), inhibitor (20 nM), or inhibitor control (20 nM) was transfected into PBMCs using Lipofectamine 2000 (Invitrogen) under serum-free conditions. Six hours later, the cells were incubated in the Th17-polarizing conditions described above. Sixty-six hours later, the cells and culture supernatants were harvested for further flow cytometry analysis, RT-PCR, and ELISA.
Sequences of miR-326 Mimics, miR-326 Inhibitors, and Their Controls
MiR-326, microRNA-326.
Statistical analysis
SPSS 22.0 software (SPSS, Inc., Chicago, IL) was used to perform statistical analysis. Normally distributed data are shown as mean ± standard deviation (M ± SD), and the comparisons were performed with Student's t-tests or one-way analysis of variance. Abnormally distributed data are displayed as the median and interquartile intervals, and the distributions between groups were compared with the nonparametric Mann–Whitney U test. Paired parametric analyses were conducted with the Wilcoxon matched pair test. Correlations between variables were determined with Pearson correlation coefficients. p-Values <0.05 were considered to be statistical significance.
Results
The demographic data and clinical features of the enrolled 113 participants (58 HT patients and 55 NC) are shown in Table 3. The two groups were matched in terms of age, sex, and BMI. No significant difference in fT3 and fT4 concentrations between the two groups was found (both p > 0.05), but the levels of TPOAb, TgAb, and TSH were significantly elevated in the HT group (all p < 0.05).
Demographic and Clinical Characteristics of Hashimoto's Thyroiditis Patients and Normal Controls
BMI, body mass index; fT3, free triiodothyronine; fT4, free thyroxine; HT, Hashimoto's thyroiditis; NC, normal controls; TgAb, antithyroglobulin antibody; TPOAb, thyroperoxidase antibody; TSH, thyrotropin; IQR, interquartile range.
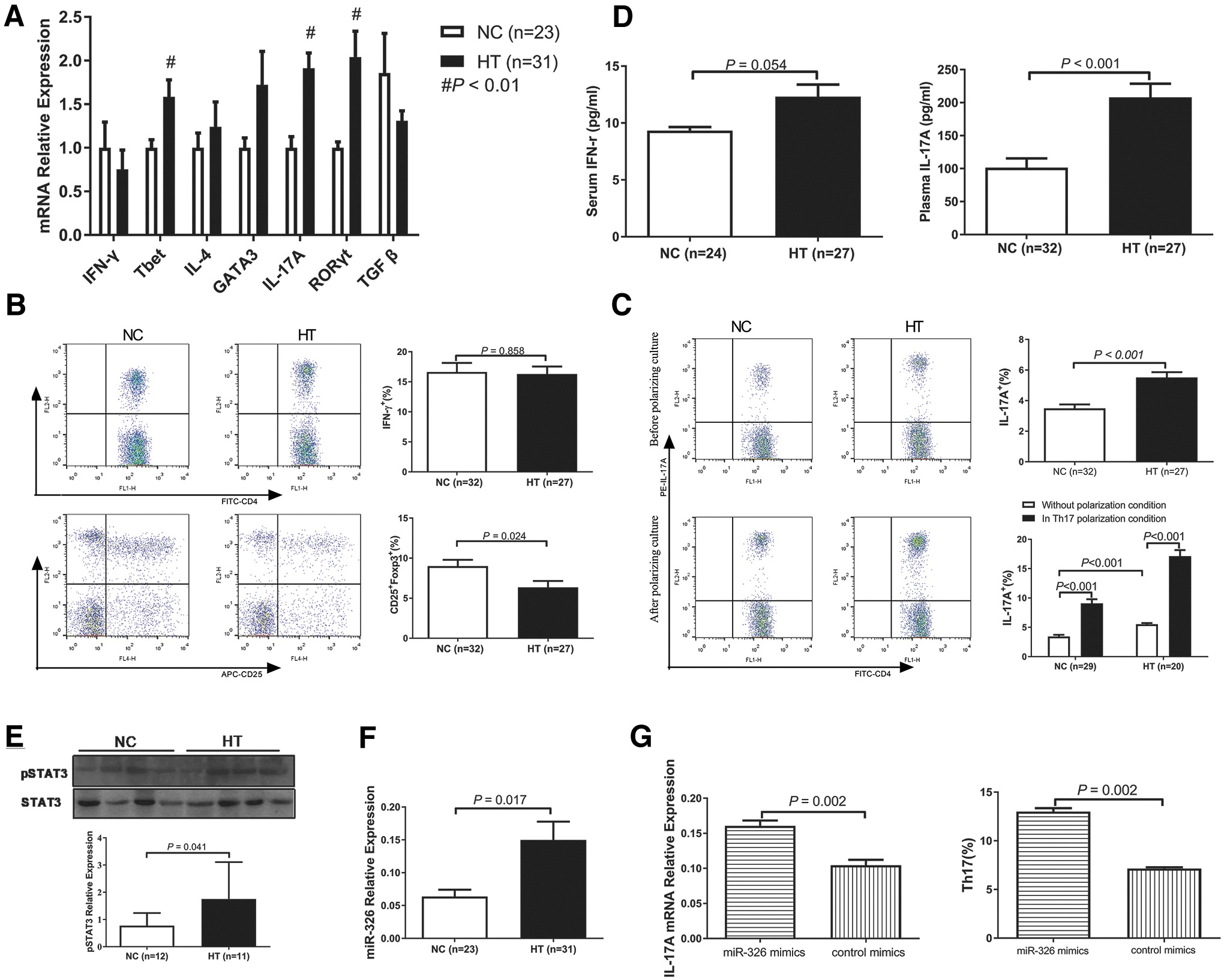
The proportion of Th17 cells is significantly elevated in HT patients
We first detected an increased mRNA expression of stereotypical cytokines and transcription factors for Th17 cells (IL-17A and RORγt, both p < 0.01), but not for Th2 cells, in the PBMCs of the HT group compared with that in the PBMCs of the NC group (Fig. 1A). Although the mRNA expression of T-bet, the transcription factor present in Th1 cells, was significantly elevated in the HT group, no difference of IFN-γ mRNA levels was found (p > 0.05). As shown by Pearson correlation analysis, IL-17A mRNA was positively correlated with both TgAb (r = 0.561 and p < 0.001) and TPOAb (r = 0.377 and p = 0.005) levels. Like IL-17A, RORγt mRNA expression was also positively correlated with both TgAb (r = 0.394 and p = 0.004) and TPOAb (r = 0.433 and p = 0.001) levels.
Elevated microRNA-326 promotes Th17 cell differentiation. (
Increased proportions of IL-17A-secreting Th17 cells (p < 0.001) and decreased proportions of CD4+CD25+Foxp3+ Treg cells (p = 0.024) in HT patients compared with those in the NC group were observed by flow cytometry, but there was no difference in the proportion of IFN-γ-secreting Th1 cells between the two groups (p = 0.858, Fig. 1B, C). The proportion of Th1 cells was not correlated with TgAb or TPOAb levels (both p > 0.05). In contrast, the proportion of Th17 cells was positively correlated with TgAb (r = 0.543 and p < 0.001) and TPOAb (r = 0.313 and p = 0.016) levels. The proportion of Treg cells was negatively correlated with TgAb (r = −0.370 and p = 0.016), but not with TPOAb (r = −0.095 and p = 0.550), levels.
We also performed ELISA to determine cytokine concentrations in serum/plasma. Concordant with the results above, plasma IL-17A levels were significantly higher in the HT group than in the NC group (p < 0.001), but this was not the case for serum IFN-γ levels (p = 0.054, Fig. 1D). To verify the enhanced number of Th17 cells in HT patients, Western blotting was performed to determine the level of pSTAT3. PBMCs derived from HT patients contained significantly more (p = 0.041) pSTAT3 (Tyr705) than those derived from the NC group (Fig. 1E).
Elevated miR-326 contributes to Th17 cell differentiation
We further detected miR-326 expression in HT patients, which was significantly higher than that in the NC group (p = 0.017, Fig. 1F). As shown by Pearson correlation analysis, miR-326 was positively correlated with the levels of both TgAb and TPOAb (r = 0.385, p = 0.007 for TgAb and r = 0.339, p = 0.020 for TPOAb). Moreover, a positive correlation between miR-326 transcripts and pSTAT3 protein levels was also found (r = 0.298, p = 0.186).
Based on these results, we wondered whether miR-326 participates in the pathogenesis of HT by regulating Th17 cell differentiation, similar to the results of our previous report in mouse models (9). To this end, we transfected PBMCs derived from HT patients with miR-326 mimics. After transfection and culture in Th17-polarizing conditions, IL-17A mRNA expression (p = 0.002) and the proportion of Th17 cells (p = 0.002) were both remarkably increased in the group transfected with miR-326 mimics compared with those in the mimic control group, as shown in Figure 1G. These results suggest that miR-326 takes part in the pathogenesis of HT, probably by regulating Th17 cells.
We then analyzed Th17 cell proportions in PBMCs after their incubation in Th17 cell-polarizing conditions. The proportion of Th17 cells increased after 72 hours of incubation (p < 0.001) but was more pronounced in the HT group than that in the NC group (Fig. 1C). These data suggest that Th17 differentiation was enhanced in HT patients compared with that in healthy controls.
CD4+ and CD8+ T cell responses to IL-23 are enhanced in HT patients
The levels of pSTAT3, which indicates a response to IL-23 stimulation (24), were determined by flow cytometry. At baseline, a higher mean fluorescence intensity (MFI) of pSTAT3 in the CD4+ T cells of HT patients was detected compared with controls (p = 0.020), but no difference in pSTAT3 in CD8+ T cells between the two groups was found (p > 0.05, Fig. 2A). Following stimulation with IL-23, the MFIs of pSTAT3 in both the CD4+ and CD8+ T cells of HT patients were higher than those of the controls (p = 0.003 and p = 0.002, respectively), and the MFI of pSTAT3 was significantly higher than the baseline level in both CD4+ and CD8+ T cells (Fig. 2A). Moreover, the fold change (Fc) for the MFI of pSTAT3 in CD4+ cells was substantially higher than that in CD8+ T cells, and the Fc in the CD4+ T cells of HT patients was much higher than that of the controls (Fig. 2B). These data suggest that CD4+ T cells from HT patients are hyper-responsive to IL-23, which may largely explain the increased differentiation into Th17 cells.

IL-23 substantially increases pSTAT3 levels in HT patient-derived PBMCs. (
Surface IL-23R expression correlates with IL-23-induced STAT3 phosphorylation
Because of the critical role of IL-23R in the IL-23/IL-23R/Th17 cell axis described above, we next determined IL-23R expression in PBMCs. As shown in Figure 3A, IL-23R protein levels were significantly higher in the PBMCs from HT patients than in those from controls (p = 0.005), although this increase was restricted to CD4+ T cells (p = 0.003, Fig. 3B). The MFI of mIL-23R in CD4+ T cells was positively correlated with the MFI of IL-23-induced pSTAT3 in both controls and HT patients (r = 0.539, p = 0.001 and r = 0.468, p = 0.014, respectively, Fig. 3C). Based on these results, we conclude that the extreme response of HT patients to IL-23 is dependent, at least in part, on increased surface IL-23R expression.
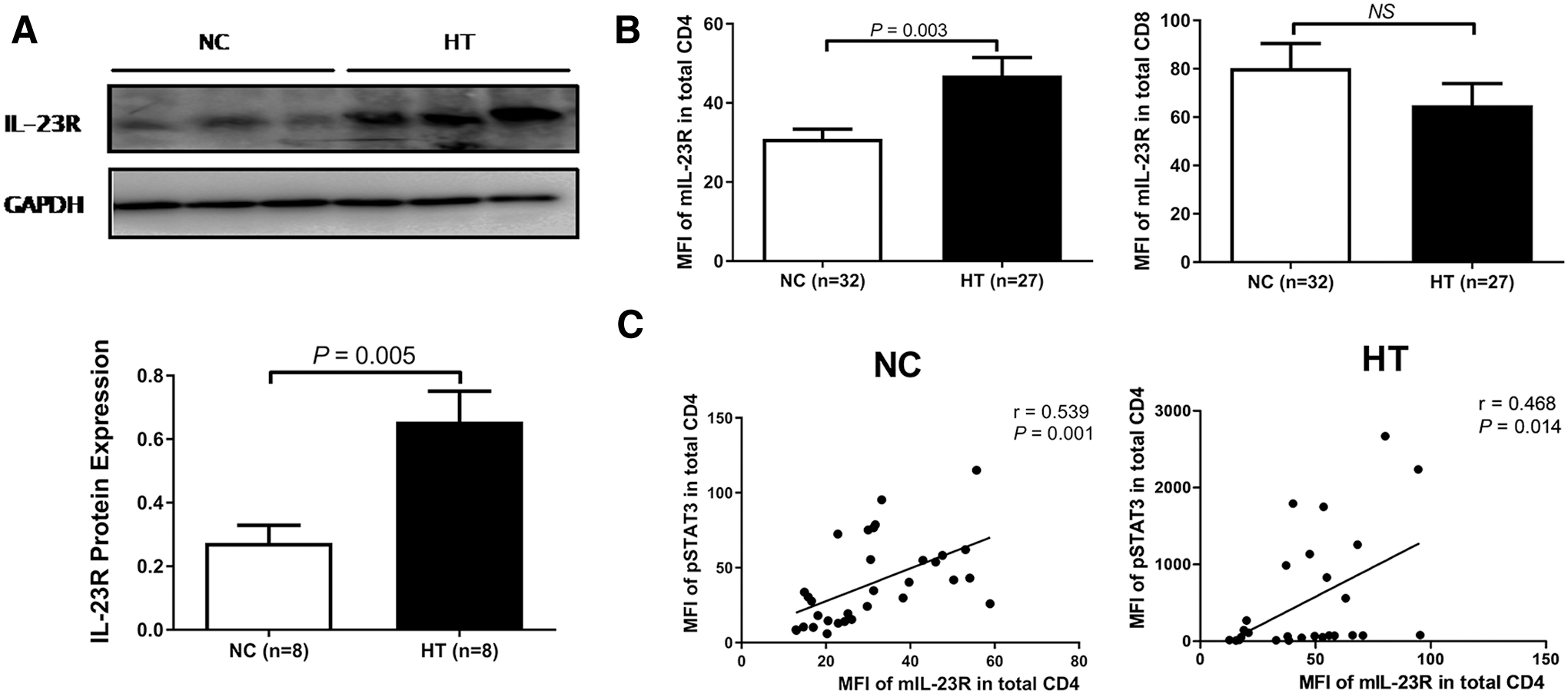
Surface IL-23R levels correlate with the MFI of IL-23-induced pSTAT3. (
miR-326 functionally targets ADAM17
Having addressed the critical role of miR-326 in Th17 cell development, we sought to gain more insight into the potential mechanism. Although ADAM17 has been reported to be a target of miR-326 in lung adenocarcinoma (21), a relationship between ADAM17 and miR-326 in HT patients has not yet been reported. To investigate a possible relationship, an miR-326 inhibitor and mimics were transfected into PBMCs derived from HT patients and healthy controls, respectively. The inhibition of endogenous miR-326 significantly increased ADAM17 protein expression (p < 0.001), but ADAM17 mRNA expression showed only a trend toward an increase (p = 0.064, Fig. 4A). In addition, the increased shedding of sIL-23R but decreased IL-17A mRNA expression and IL-17A protein expression in the cell supernatant were observed in transfected cells compared with that in cells transfected with the inhibitor control (all p < 0.05, Fig. 4B). In contrast, transfection with miR-326 mimics significantly attenuated ADAM17 expression and shedding, but enhanced surface IL-23R and IL-17A expression (p < 0.05). These data suggest that miR-326 regulates ADAM17 at both the mRNA and protein levels.
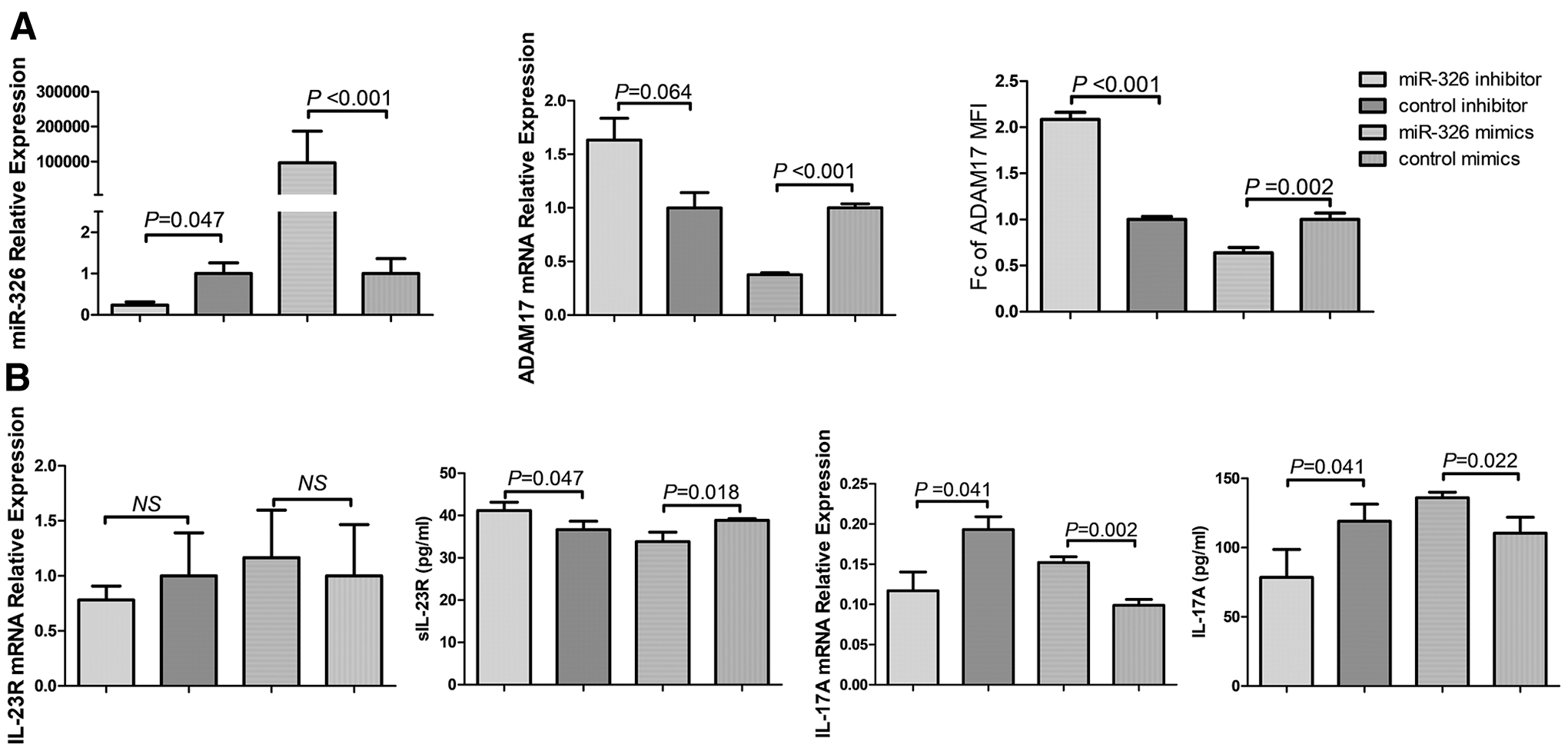
ADAM17 is a target of miR-326. (
Inhibition of ADADM17 expression may increase surface IL-23R expression
We next determined the expression of ADAM17 in HT patients. As shown in Figure 5A, the expressions of both mRNA and protein were significantly decreased in HT patients relative to those in controls (p = 0.003 and p = 0.010, respectively). Consistently (Fig. 5B), surface ADAM17 levels in both the CD4+ and CD8+ T cells of the HT patients were higher than those of the controls (p = 0.001 and p < 0.001, respectively). Moreover, the ADAM17 mRNA level was negatively correlated with the TgAb and TPOAb levels (r = −0.346, p = 0.012 for TgAb; r = −0.332, p = 0.016 for TPOAb). In addition, there was a negative correlation between ADAM17 protein expression and TgAb levels (r = −0.468, p = 0.033). A similar trend between ADAM17 protein expression and TPOAb levels (r = −0.405, p = 0.068) was found, although this correlation was not significant.

Surface IL-23R is a substrate of ADAM17. (
ELISA was used to explore the shedding of sIL-23R, which may antagonize surface IL-23R, in plasma. Compared with that in controls, as shown in Figure 5C, a significant reduction in sIL-23R shedding was observed in HT patients (p < 0.001). In addition, plasma sIL-23R concentrations were positively correlated with the MFI of ADAM17 in CD4+ T cells (r = 0.431, p = 0.001).
To verify the ectodomain shedding of surface IL-23R by ADAM17, purified CD3+ T cells isolated from HT patients were incubated with TAPI-1, which inhibits ADAM17. Then, surface ADAM17 and IL-23R expression was analyzed by flow cytometry. As shown in Figure 5D, surface ADAM17 levels were elevated with T cell receptor activation (p = 0.005), but the shedding of surface IL-23R was arrested in the presence of TAPI-1 (p = 0.035). Taken together, these data indicate a mechanistic link between miR-326 and the IL-23/IL-23R/Th17 cell axis in HT patients, which is enhanced by the targeting of ADAM17 by miR-326.
Discussion
In the present study, we demonstrate that the miR-326-induced differentiation of Th17 cells through ADAM17 targeting may be involved in the pathogenesis of HT. Recent research reported that elevated Th17 cells play critical roles in the pathogenesis of HT, which was once attributed to abnormal proportions of Th1 cells (4,25). A previous study in a mouse model of HT has shown that an extreme high iodine intake may induce Th1 cell polarization, but the differentiation of Th17 cells could be facilitated by moderate iodine intake (26). All the individuals in this study resided in an area with adequate iodine levels (27), which may partially explain the increased Th17 cells, but not Th1 cells, in HT patients. In addition, the study by Nanba et al. has reported that the Th17 cell proportions in HT patients, regardless of thyroid function, were higher than those in healthy controls, but the proportions of Th1 cells in HT patients with hypothyroidism were significantly higher than those in euthyroid HT patients (28). In this respect, an alternative explanation is that the disease was not severe enough because about 66.7% of the enrolled HT patients were euthyroid.
The enhanced generation of Th17 lymphocytes when CD4+ T cells are cocultured with autologous monocyte-derived dendritic cells or cultured in Th17-polarizing conditions has been observed in patients with rheumatoid arthritis (29). Upon IL-23 stimulation, HT-derived PBMCs produced more Th17 cells (14). Consistent with these reports, our results indicate that more Th17 cells are produced after incubation under Th17-polarizing conditions and that this phenomenon is more pronounced in HT patients than in NC. These data suggest that PBMCs from HT patients have a greater potential to differentiate into Th17 cells. Binding with IL-23R, IL-23 plays a critical role in the proliferation and stabilization of Th17 cells, and IL-23R surface expression is the major determinant in the IL-23/IL-23R signaling system (30). In this study, elevated mIL-23R levels detected in HT patients were correlated with the enhanced response to IL-23. Based on these data, it is tempting to propose that elevated IL-23R surface expression in CD4+ T cells explains, at least in part, the increased differentiation of HT-derived PBMCs in Th17 cells.
Abnormal miR-326 expression has been reported in a variety of autoimmune diseases (31). In the present study, we detected increased miR-326 levels in HT patients, which was consistent with our previous study in animal models (9). With the overexpression of miR-326 in HT patient-derived PBMCs, the differentiation of Th17 cells was enhanced. These results suggest a critical role for miR-326 in the regulation of Th17 cells in HT patients. ADAM17 is a target of miR-326 and participates in cell invasion in lung adenocarcinoma (21). In our study, HT patients displayed decreased ADAM17 expression at both the mRNA and protein levels. Mechanistic studies detected elevated ADAM17 mRNA and protein levels in HT-derived PBMCs transfected with the miR-326 inhibitor and vice versa in healthy control-derived PBMCs. Based on these data, we propose that increased miR-326 may target ADAM17 to participate in HT pathogenesis.
With advances in research, IL-23-neutralizing agents have been shown effective in treating several autoimmune diseases. Parham et al. first reported that the human IL-23R complex is a type I transmembrane protein comprising 629 amino acids (32). In this study, mIL-23R expression in HT patients was significantly higher than that in the control group, which was consistent with previous studies (14,33). Recent mechanistic studies confirmed that sIL-23R, a cytokine-binding homology region, acts as an extracellular receptor analogue to compete with IL-23 and inhibit the differentiation of Th17 cells through inhibiting the STAT3 signaling pathway (34). Kan et al. (35) detected 24 different IL-23Rα mRNA transcripts, which may alter IL-23Rα protein expression. The authors then discovered that Δ9, an alternative splice form of IL-23Rα mRNA, encodes a soluble form of the IL-23R extracellular domain that antagonizes full-length IL-23R, blocks IL-23/IL-23R signaling, and inhibits the generation of Th17 cells, but the content of this variant in human peripheral blood was low (36).
sIL-23R can be obtained from the translation of spliced mRNA or protein posttranslational modification. Ectodomain shedding of mIL-23R was identified as an alternative pathway for the generation of sIL-23R. A recent study reported that sIL-23R, which acts as a natural antagonist of IL-23/IL-23R signaling, is derived from mIL-23R by the shedding activity of ADAM17 (18). An investigation in type 1 diabetes mellitus found that membrane IL-6R can be cleaved by the protease ADAM17 and that this cleavage may block IL-6/IL-6R signaling and inhibit inflammation (23). In this study, plasma sIL-23R levels in HT patients were lower than those in the control group, while the levels of mIL-23R were higher, suggesting that sIL-23R originates from shedding of the extracellular segment of mIL-23R. Based on the correlation between sIL-23R concentration and ADAM17 protein expression, we hypothesize that enhanced sIL-23R may be due, at least in part, to the cleavage of mIL-23R by ADAM17. When ADAM17 was inhibited, mIL-23R expression increased significantly, and sIL-23R levels in the cell supernatant decreased, further confirming the effect of ADAM17 in shearing mIL-23R. Low levels of ADAM17 in HT patients resulted in a relative increase in mIL-23R levels, which may explain the enhanced response of HT patients to IL-23 stimulation. These results suggest that ADAM17 antagonizes IL-23/IL-23R signaling in HT patients, which provides a new target for the treatment of HT in the future.
Our study has some limitations. First, ADAM17 is not the only target of miR-326, and hence, we were unable to absolutely exclude alternative mechanisms by which miR-326 regulates Th17 cells. Second, the targeting of ADAM17 by miR-326 has not been proven in a luciferase reporter assay in the present study, although this has been performed in a previous study (21). Finally, mechanistic studies were conducted ex vivo but not in vivo, which would be more definitive. Further in vivo studies are needed to corroborate the findings.
In summary, we found an enhanced IL-23/IL-23R/Th17 cell axis in HT patients, which may be attributed, at least in part, to the targeting of ADAM17 at both the RNA and protein levels by elevated miR-326.
Footnotes
Acknowledgments
We are grateful to the participants of this study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by the National Natural Science Foundation of China (grant numbers 81471003).