
Abstract
Resistance to thyroid hormone alpha (RTHα) is a rare and under-recognized genetic disease caused by mutations of THRA, the gene encoding thyroid hormone receptor α1 (TRα1). We report here two novel THRA missense mutations (M259T, T273A) in patients with RTHα. We combined biochemical and cellular assays with in silico modeling to assess the capacity of mutant TRα1 to bind triiodothyronine (T3), to heterodimerize with RXR, to interact with transcriptional coregulators, and to transduce a T3 transcriptional response. M259T, and to a lower extent T273A, reduces the affinity of TRα1 for T3. Their negative influence is only reverted by large excess of T3. The severity of the two novel RTHα cases originates from a reduction in the binding affinity of TRα1 mutants to T3 and thus correlates with the incapacity of corepressors to dissociate from TRα1 mutants in the presence of T3.
Introduction
The currently known germline heterozygous mutations in THRA, which encodes thyroid hormone receptor α1 (TRα1), result in many signs of hypothyroidism. The genetic disease, called resistance to thyroid hormone alpha (RTHα), is notably associated with altered skeletal growth and cognitive functions, with near-normal thyroid function tests (1). However, based on the report of only 24 cases, its symptoms and severity seem to be quite variable (2). We report here two patients with germline heterozygous novel missense mutations in THRA causing RTHα.
Results
Two novel cases of RTHα
Two cases of RTHα were diagnosed in two hospitals in France (Table 1; Methods section in Supplementary Data). Case A was a girl born prematurely, with a twin brother. The mother had medically assisted reproduction. The girl was immediately hospitalized due to transient respiratory distress, severe constipation with megacolon, and malaises due to reflex vagal hypertonia. Macroglossia was first noted at 4 months. She had an operation for bilateral ovarian hernia. Her motor development was delayed, and she was unable to sit for the first year. She could walk only at three years old. In contrast, language acquisition was normal from 1 to 3 years old. Mild hypotonia and a coarse voice were noticed at two years old.
Clinical and Biochemical Parameters of Patients
Also present in unaffected family members.
Measured without interruption of levothyroxine treatment.
BMI, body mass index; fT3, free triiodothyronine; fT4, free thyroxine; NA, not available; SD, standard deviation; T3, triiodothyronine; T4, thyroxine; TSH, thyrotropin.
Brain magnetic resonance imaging (MRI) showed periventricular high signals, compatible with a myelination defect, but no morphological anomaly. In particular, cerebellum foliation was normal. By the age of three years, she developed regression in terms of language, communication, and playing. At four years old, she had anxiety and depression, repetitive behavior, and fatigue. Chronic constipation was also noticed. Neurologic examination was normal with the exception of a discreet enlargement of polygona and mild global hypotonia. Interestingly an excess of several amino acids was measured in serum and urine at this point, but metabolic investigations were not continued (Table 1).
Between 5 and 9 years old, the patient had many stereotyps, anxiety attacks, and performed automutilation. Her gait remained slightly unstable and skeletal growth was slow. Epileptic seizures occurred between age 8 and 11 years. At age 14 years, because of frequent nausea and vomiting, lack of appetite and weight loss, she had tube feeding. Cold sensitivity was also reported.
While thyroxine (T4), triiodothyronine (T3), and thyrotropin (TSH) levels remained within normal range in the patient, the T3 level was in the upper range, and a low ratio between fT4 to free triiodothyronine (fT3) was noted (from 3.4 to 1.6), which was a clue for RTHα (2). Exome sequencing identified a THRA mutation (c.776T>C;M259T) and
Case B was a girl of Guinean origin, born through cesarean section. She was first examined at 2 years old for psychomotor retardation. She was unable to speak, and had an unstable gait. This was associated with macrocephaly and obesity (Table 1). Brain MRI was normal. fT4/fT3 ratio was low (1.4). TSH responded normally to a thyrotropin-releasing hormone challenge (protirelin 250 μg; Ferring France). The concentrations of other pituitary hormones were normal. She did not display any delay in skeletal growth, and the insulin-like growth factor 1 (IGF1) level was within normal range. Developmental support was initiated at 2 years old, together with
Targeted sequencing was performed for genes involved in T3 signaling when the patient was 13 years old. The patient was found to have a heterozygous THRA mutation (c.817A>G; T273A) absent in her mother and three of her siblings. Other family members could not be genotyped. Except for macrocephalia, which was present in her mother and all her brothers and sisters (58–60 cm +2 to +3 standard deviation), other family members did not display any symptoms of RTHα.
Functional characterization of mutant receptors in transfected cells
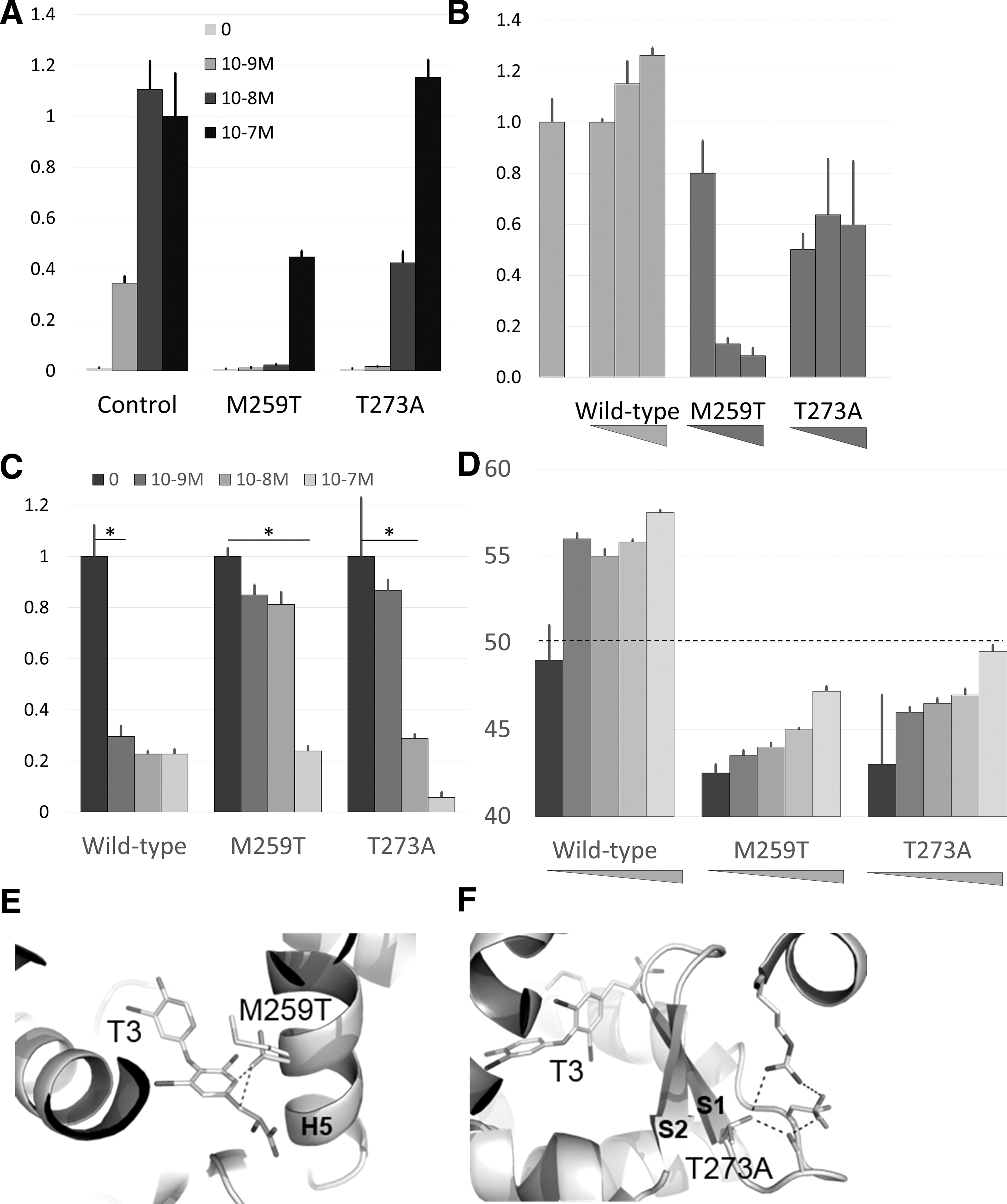
An expression vector for wild-type or mutant TRα1 was transfected into HEK293T cells, together with a reporter construct with a minimal promoter containing two DR4 response elements. While the mutations did not affect TRα1 stability in transfected cells (Supplementary Fig. S1), they significantly reduced its transactivation capacity (Fig. 1A). We then mixed a fixed amount of the DNA vector for wild-type TRα1 expression, with increasing amounts of mutant TRα1, to assess the dominant negative activity of mutant TRα1 in the presence of T3 (10−8 M). In this setting, TRα1M259T and, to a lesser extent, TRα1T273A reduced the T3 response of the cells (Fig. 1B).
(
As the mechanism underlying the dominant negative influence of mutant TRα1 is thought to reflect its incapacity to dissociate from corepressors in the presence of T3 (2), we used a two-hybrid assay to evaluate the influence of TRα1 mutations on its interaction with nuclear receptor corepressor (NCoR). A VP16 activation domain was appended at the N-terminus of TRα1, and the NCoR nuclear interaction domain, including CoRNR1 and CoRNR2 motifs, was fused to the DNA binding domain of the Gal4 transcription factor. This assay revealed that TRα1M259T failed to dissociate from NCoR, even at very high concentration of T3, while the dissociation of TRα1T373A was observed only at high T3 concentration (Fig. 1C).
Biochemical characterization of the ligand binding domain of TRα1 mutants
We produced, in Escherichia coli, recombinant proteins corresponding to wild-type and mutants TRα1 ligand binding domains (LBDs). We verified their purity and structural integrity using sodium dodecyl sulfate page and circular dichroism (Supplementary Fig. S2). The structural stability of LBDs was then assessed in thermal shift assays that monitor the fraction of unfolded protein as a function of temperature (Fig. 1D). Differential melting temperature measurements revealed a destabilization of the unliganded TRα1 LBD by the M259T (−7.2°C) and T273A (−6.4°C) substitutions. T3 addition induced only a slight stabilization of mutant LBDs, consistent with a strong decrease in the T3 affinity for the TRα1 mutants. The loss of affinity for T3 was confirmed by isothermal titration calorimetry (Supplementary Fig. S3).
Inspection of the structure of the wild-type TRα1 LBD bound to T3 (Protein Data Bank [PDB] code 2H79) (3) provided a likely explanation for these defects. This showed that the linear residue M259 makes favorable van der Waals interactions with T3, whereas the presence of a branched threonine at this position is very likely to create steric clashes with T3 and surrounding residues (C255, M256) (Fig. 1E). The structure also showed that the hydroxyl group of T273 is involved in a hydrogen bond network stabilizing the β-sheet S1–S2, which is part of the ligand binding pocket (Fig. 1F). Its substitution into an alanine breaks this network.
As predicted, the mutations did not alter the capacity of TRα1 to form heterodimers with RXRα (Supplementary Fig. S4). Fluorescence anisotropy measurements also showed that the mutation did not alter the capacity of the unliganded TRα1 LBD to recruit fluorescent peptides derived from the NCoR corepressor (CoRNR1 and CoRNR2 motifs; Supplementary Table S1; Supplementary Fig. S5) and to associate, when a large excess of T3 is present, with a peptide derived from the SRC-1 coactivator (NR2 motif; Supplementary Table S1; Supplementary Fig. S5).
Discussion
The symptoms of patient A and B are consistent with previous reports (1) and emphasize mental retardation as a common trait. However, the skeletal growth of patient A was initially normal, without thyroxine supplementation, and only patient B had macrocephalia. Other frequent symptoms of RTHα, like anemia, and the presence of skin tags, were absent. The excess of amino acids in the serum of patient A may be circumstantial. However, a recent metabolomics analysis of mouse models raises the possibility that these could be serum markers of the disease (4).
The mutations found in the two patients have equivalent mutations in RTHβ patients [TRβ1M313V (5) and TRβ1T327A (6)]. They modify the structure of the ligand binding pocket and reduce the affinity for T3. In agreement with another recent study (7), a clear correlation exists between the severity of the symptoms and the inability of T3 to dissociate NCoR from the mutant receptors, and the resulting dominant negative activity exerted by these TRα1 variants in heterozygous cells.
Footnotes
Acknowledgments
We thank Chloé Morin for technical help in plasmid construction and transfection experiments. We thank Caroline Mas (ISBG, Grenoble) for technical assistance for ITC experiments. We acknowledge the use of OMNISEC equipment of Laboratory of Spectroscopy and Calorimetry (LEC) at Brazilian Biosciences National Laboratory (LNBio), CNPEM in Campinas.
Author Disclosure Statement
The authors have nothing to disclose.
Funding Information
M.P. was supported by the French National Research Agency (ANR-14-ACHN-0016). This study was supported by a grant from Agence Nationale de la Recherche (Thyromut2 program; ANR-15-CE14-0011-01). The CBS is a member of the France-BioImaging (FBI) and the French Infrastructure for Integrated Structural Biology (FRISBI), supported by the French National Research Agency (ANR-10-INBS-04-01 and ANR-10-INBS-05, respectively). The authors acknowledge the support and the use of resources of Instruct-ERIC and the financial support from the CNPq Programa Ciencia Sem Fronteiras (BJT 300143/2015-0), the CNPq Programa Universal (420416/2016-1).
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5