
Abstract
Background:
Hashimoto's thyroiditis (HT) is a common autoimmune disease of unknown origin. However, viral infections have been implicated as triggers for autoimmunity. Human leukocyte antigen (HLA) class I presents antigens to circulating immune cells and plays a crucial role in the defense against viral infections. This study aimed to investigate the presence of enterovirus and HLA class I expression in one of the largest HT thyroid tissue cohorts to date. In addition, viral receptors and viral immune response proteins were examined.
Methods:
Thyroid tissue samples from 46 HT patients were obtained using core needle biopsy. Thyroid tissue collected during neck surgery for other reasons than thyroid autoimmunity served as controls. Standard immunohistochemistry on formalin-fixed, paraffin-embedded tissue samples were used to detect HLA class I, enteroviral capsid protein 1 (VP1), and coxsackie and adenovirus receptor (CAR) in thyroid cells. A subset of the samples was examined with combined immunofluorescence staining for signal transducer and activator of transcription 1 (STAT1) and protein kinase R (PKR).
Results:
Significantly more HLA class I-positive samples were found in the HT group (31 out of 46 [67.4%]) than in the control group (5 out of 24 [20.8%]) (p < 0.001). Moreover, the semiquantitative score assessing the grade of HLA class I expression was significantly higher in the HT group (3.9 ± 3.1) than in the control group (0.5 ± 0.9) (p < 0.001). In addition, STAT1 was colocalized with HLA class I, and PKR and VP1 were also found and were colocalized together. VP1 was detected in both controls and the HT samples, with slightly more VP1+ thyroid cells in the HT samples (20.1% ± 16.4%) than in controls (14.9% ± 10.5%). Finally, the presence of CAR in thyroid cells was confirmed.
Conclusion:
The current study confirmed that HLA class I hyperexpression is a defining feature of HT. Thyroid cells express CAR, thus making them susceptible to enterovirus infection. The colocalization of HLA class I with STAT1 and VP1 with PKR indicates an intracellular, antiviral host response. These findings support the concept of a firm link between viral infection and autoimmune thyroid diseases.
Introduction
Hashimoto's thyroiditis (HT) is a common autoimmune disease characterized by the generation of autoantibodies to thyroglobulin and thyroid peroxidase, lymphocytic infiltration of the thyroid gland, and for some patients, overt hypothyroidism. A strong genetic component has been established (1,2), implicating both thyroid-specific susceptibility genes and genes involved in immunoregulatory mechanisms (3,4). In addition to genetic influences, environmental factors are also believed to play an important part in the development of HT. Despite this, the etiology of HT remains unknown, and the immunological pathway leading to the characteristic lymphocytic displacement of thyroid cells has yet to be fully described. Evidence of an association between autoimmune thyroid diseases (AITD) and viral infections is, however, emerging (5 –12). Still, the evidence linking viral infection to disease development is limited and direct investigation of thyroid tissue for evidence of antiviral responses has rarely been undertaken. Nevertheless, an immunological thyroid tissue response similar to that seen in viral infection has previously been described in the current cohort of 46 HT patients (13). Therefore, this study aimed to augment these findings by further investigation of viral receptors, antigen presentation, and viral immune response proteins.
Coxsackie and adenovirus receptor (CAR) is a transmembrane glycoprotein that serves as the main cellular entry route for Coxsackie B virus and adenovirus. In addition, more complex functions involving endothelial tight junction regulations and epithelial permeability have been identified (14,15). Four of five known isoforms of CAR bind enterovirus, but only two of the isoforms (CAR-SIV and CAR-TVV) contain a transmembrane domain, thus making them able to mediate an intracellular infection. RNA sequencing studies recently demonstrated that the SIV-isoform was the most highly expressed CAR isoform in the thyroid (16).
The major histocompatibility complex (MHC) class I, also called the human leukocyte antigen (HLA) class I in humans, plays a crucial role in the defense against viral infections. Once a virus enters a cell, peptides derived from virally synthesized proteins are presented by HLA class I on the cell surface to circulating CD8+ T cells. This leads to an immune response resulting in destruction of the infected cell. Although all nucleated cells express HLA class I, the molecule complex can only be stably expressed on the cell surface when loaded with peptide (17). Additionally, its expression can be affected by viral infections, with some viruses downregulating HLA class I expression, while others cause an upregulation of HLA class I expression (18 –25).
Signal transducer and activator of transcription 1 (STAT1) and protein kinase R (PKR) are antiviral proteins activated by interferons and the viral product dsRNA, respectively (26 –29). Upon activation by interferons, STAT1 translocates from the cytoplasm to the nucleus and initiates transcription of antiviral interferon-stimulated genes (30 –33). Indeed, one of the interferon-stimulated genes transcribed in response to activated STAT1 is PKR. PKR modifies and interacts with STAT1, although the effects of PKR on STAT1 are not fully understood. Nevertheless, studies have shown that PKR-deficient cells display reduced and defective STAT1-dependent functions, including transactivation and interferon-stimulated phosphorylation of STAT1 (34). Interestingly, STAT1 activation has been found to underpin HLA class I upregulation by virally induced interferons.
We previously reported an increased expression of the downstream interferon type 1 response protein, myxovirus resistance protein 1 (MxA), as well as increased numbers of CD8+ T cells and plasmacytoid dendritic cells in this collection of HT thyroid tissue and in Graves' disease (13,35). Furthermore, enteroviral capsid protein 1 (VP1), which is found in all enteroviruses, have been reported in thyroid cells in both Graves' disease (10) and in the current HT cohort (13).
On this basis, we hypothesized that thyroid cells are susceptible to enteroviral infection and that the virus elicits an immune response in thyroid tissue that triggers autoimmunity. In the current study of thyroid tissue from HT patients, our aims were to: (i) investigate the presence of CAR-receptor; (ii) assess HLA class I expression; (iii) explore the presence of STAT1 and PKR; and (iv) reinvestigate the presence of VP1. Using immunohistochemistry (IHC), we assessed the expression of CAR-SIV, HLA class I, STAT1, PKR, and VP1 in thyroid tissue from a large cohort of mainly newly diagnosed HT patients.
Materials and Methods
Study participants and thyroid tissue collection
For this study, we used previously collected tissue samples. As reported earlier, thyroid tissue from 46 patients with HT (defined as antithyroid peroxidase antibodies [TPO-Ab] >34 kIU/L with or without thyroid dysfunction) and 24 controls were collected at Oslo University Hospital (13). Age, sex, duration from diagnosis to thyroid biopsy, TPO-Ab, thyrotropin (TSH), and thyroid function tests (free thyroxine [fT4] and free triiodothyronine) were determined (Table 1). HT patients were divided into three subgroups according to thyroid function tests: overt hypothyroidism (TSH ≥10 mIU/L), subclinical, nontreated hypothyroidism (3.6 mIU/L ≤ TSH ≤10 mIU/L and fT4 8–20 pmol/L), and nontreated euthyroid thyroiditis (presence of TPO-Ab and normal thyroid function tests).
Patient Characteristics
Duration is the time from diagnosis to biopsy. Values are presented as mean ± SD, numbers (%) or median and range (duration). p-Values compared with controls and tested with Student's t-test, Mann–Whitney U test or Pearson's chi-square test.
fT4, free thyroxine; TPO-Ab, thyroid peroxidase antibodies; TSH, thyrotropin.
Tissue samples from HT patients were collected by core needle biopsy. Thyroid tissue samples from 24 patients undergoing neck surgery for other reasons than AITD, that is, thyroid tumors or parathyroid adenomas, served as controls. We assured that all controls had no pre-existing or unrecognized thyroid autoimmunity by measuring serum TPO-Ab and antibodies against TSH-receptor and thyroglobulin.
The thyroid specimens were taken from normal thyroid tissue, adjacent to the pathological lesion. The formalin-fixed, paraffin-embedded tissue samples were cut into 3 μm slices and mounted on slides for further analysis. The Regional Ethics Committee approved the study and written informed consent was obtained from all participants.
Nine (19.6%) HT patients had other autoimmune diseases (rheumatoid arthritis n = 2, celiac disease n = 2, type 1 diabetes n = 1, type 1 diabetes and celiac disease n = 1, pernicious anemia n = 1, ankylosing spondylitis n = 1, systemic lupus erythematosus, and Sjögren's syndrome n = 1). Only one HT patient received immunosuppressive therapy (steroids) before inclusion in the study. Two patients in the control group (8.3%) had an additional autoimmune disease (ulcerous colitis n = 1 and psoriasis n = 1). None of the control patients received immunosuppressive therapy before study inclusion.
Immunohistochemistry
HLA class I and VP1 immunostaining was performed with a standard IHC protocol. Antigens were unmasked by heating in 10 mM citrate buffer pH 6.0, in a pressure cooker in a microwave oven on full power (800 W) for 20 minutes, followed by 20 minutes of cooling at room temperature. Sections were blocked with 10% goat serum and primary antibodies (Supplementary Tables S1 and S2) diluted in Dako REAL antibody diluent (Agilent S202230). The Dako anti-enteroviral VP1 (5D8/1 clone) was used at a dilution of 1:1400 (55 ng/mL) for 30 minutes. The HLA class I primary antibody (Abcam class I HLA [EMR8/5] Ab70328) was used at a dilution of 1:1500 for one hour. CAR-SIV (Abcam Ab100811 antibody) was used at a dilution of 1:1000 for two hours. Primary antibodies were visualized using the Dako REAL EnVision Detection system (Agilent K5007).
Combined immunofluorescence
A subgroup of samples were immunostained (Supplementary Tables S1 and S2) sequentially with STAT1 (Ab109320, dilution 1:500, overnight), followed by HLA class I (1:1000, one hour). The same subset of cases were stained simultaneously for PKR (Abcam ab32052 at a dilution of 1:700, overnight) and VP1 [Dako anti-enteroviral VP1 (5D8/1 clone), 1:1000 dilution, overnight primary incubation]. Primary antibodies were detected using species-specific secondary antibodies conjugated to either AlexaFluor® 488 or AlexaFluor® 555 as appropriate (Invitrogen, United Kingdom).
Image acquisition and analysis
Bright-field image acquisition and analysis were performed on a Nikon 50i Microscope fitted with a DS-Fi camera and a DSL2 camera control unit. Images were captured and analyzed using the ImageJ platform. Immunofluorescence image collection and processing were achieved using a Leica DM4000 B LED upright fluorescence microscope and Leica Image analysis software (LASX).
Immunohistochemical analysis
The slides were analyzed by light microscopy on 400 × magnification by two independent scientists (T.W. and S.J.R.). First, samples were classified as positive or negative for HLA class I. Only immunostaining of thyroid follicular cells (thyrocytes) was evaluated. Next, HLA class I immunoreactivity was graded according to the semiquantitative Allred scoring system. The Allred scoring system takes both intensity (0 to 3) and proportion (0 to 5) into account, with eight being the maximum score possible and 0 being the lowest score. This system is used commonly in clinical settings to assess the immunostaining of pathological specimens (36).
We assessed VP1 staining by counting all positively stained thyrocytes alongside the total number of thyrocytes in 10 consecutive counting grids (0.058 mm2) on 400 × magnification, thus yielding a percentage of positively stained thyrocytes. The immunostaining and interpretation were both performed in a blinded fashion.
Statistics
Results are presented as mean and standard deviation, or as numbers and percentages. We calculated statistical significance of differences between mean values with the independent samples t-test if fulfilling criteria of normality, and with the Mann–Whitney U test if not normally distributed. Pearson's chi-square test was used to determine the statistical significance of differences in proportions. Associations were explored using binary logistic regression and Spearman's correlation. All analyses were performed using IBM SPSS Statistics version 25 and GraphPad Prism 7.02. We considered p-values less than 0.05 significant.
Results
Few studies have characterized the immunological responses in thyroid tissue from HT patients. In the present study, we found immunologically active HLA class I and several antiviral immune response proteins. Moreover, we confirmed that thyrocytes are susceptible to enterovirus infection.
CAR is expressed in thyrocytes
To explore whether thyrocytes might be susceptible to enterovirus infection, we analyzed the expression of CAR. We chose to examine the SIV-isoform, since this is known to mediate intracellular infection and was recently shown to be the most highly expressed CAR isoform in the thyroid by RNA sequencing (16). We found CAR-SIV in thyrocytes in all 10 samples studied. CAR-SIV had a distinct granular expression (Fig. 1A).
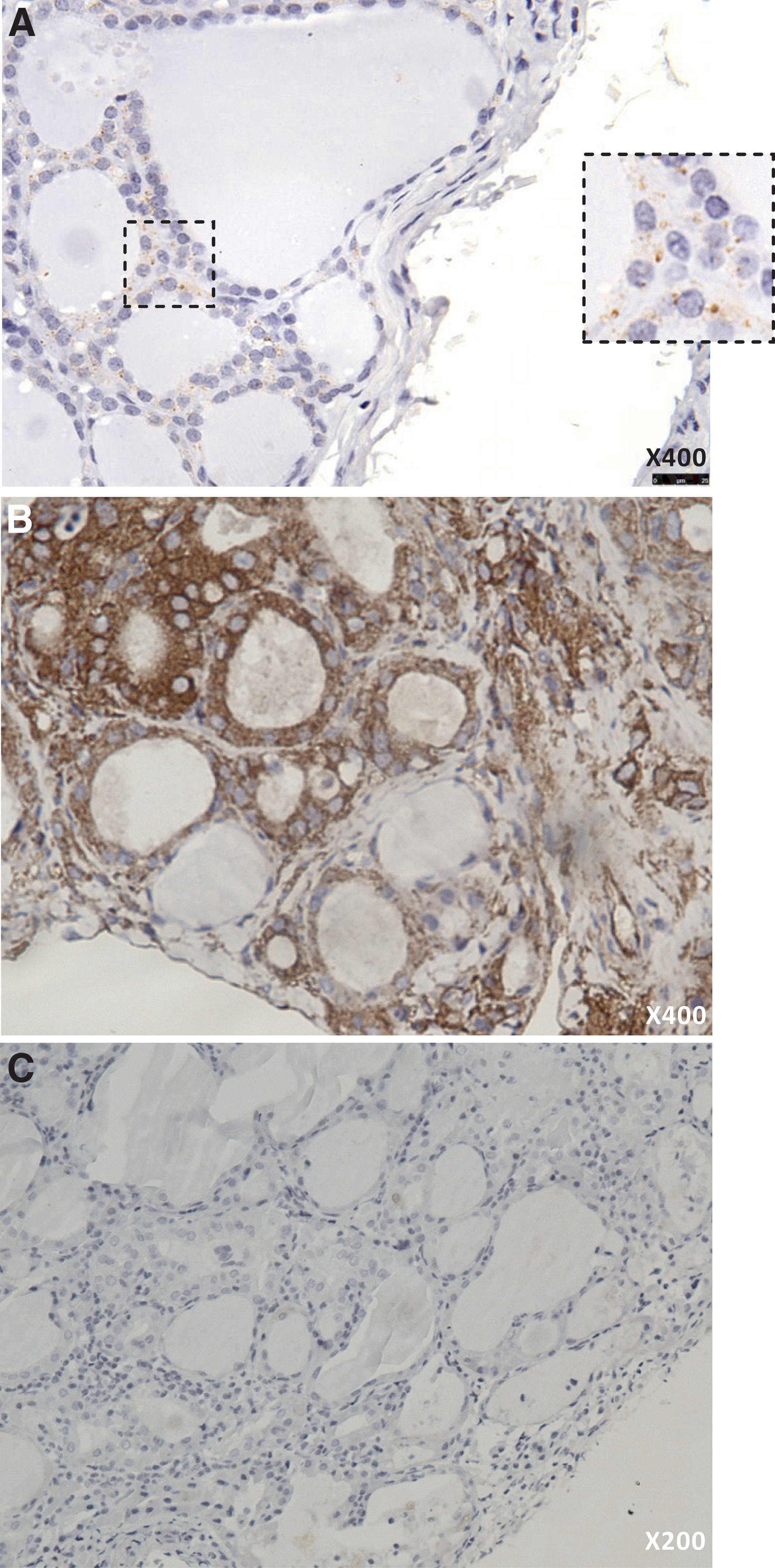
Immunohistochemical staining of CAR and HLA class I of thyroid tissue samples from HT patients and controls. CAR-SIV was found in thyrocytes of all samples studied, both HT and controls. (
HLA class I is upregulated in HT
HLA class I presents both endogenously and exogenously derived antigens to CD8+ T cells of the immune system, and plays a vital role in the body's defense against viruses. In the present study, we demonstrate that compared with controls, HLA class I is upregulated in the thyrocytes of HT patients (Fig. 1B, C). We found HLA class I+ thyrocytes in 31 out of 46 (67.4%) thyroid tissue samples in the HT group and in 5 out of 24 (20.8%) thyroid tissue samples in the control group (p < 0.001). All three patient subgroups had a significantly higher number of HLA class I-positive samples than controls. Among the subgroups, thyroid tissue from patients with overt hypothyroidism had the highest proportion of HLA class I positivity with 11 out of 15 (73.3%) being positive (Fig. 2A). In the subclinical hypothyroidism group, 9 out of 14 samples were positive (64.3%), and in the nontreated euthyroid thyroiditis group, 11 out of 17 samples were positive (64.7%) (Fig. 2A). There were no statistically significant differences between the number of HLA class I-positive samples among the three subgroups.
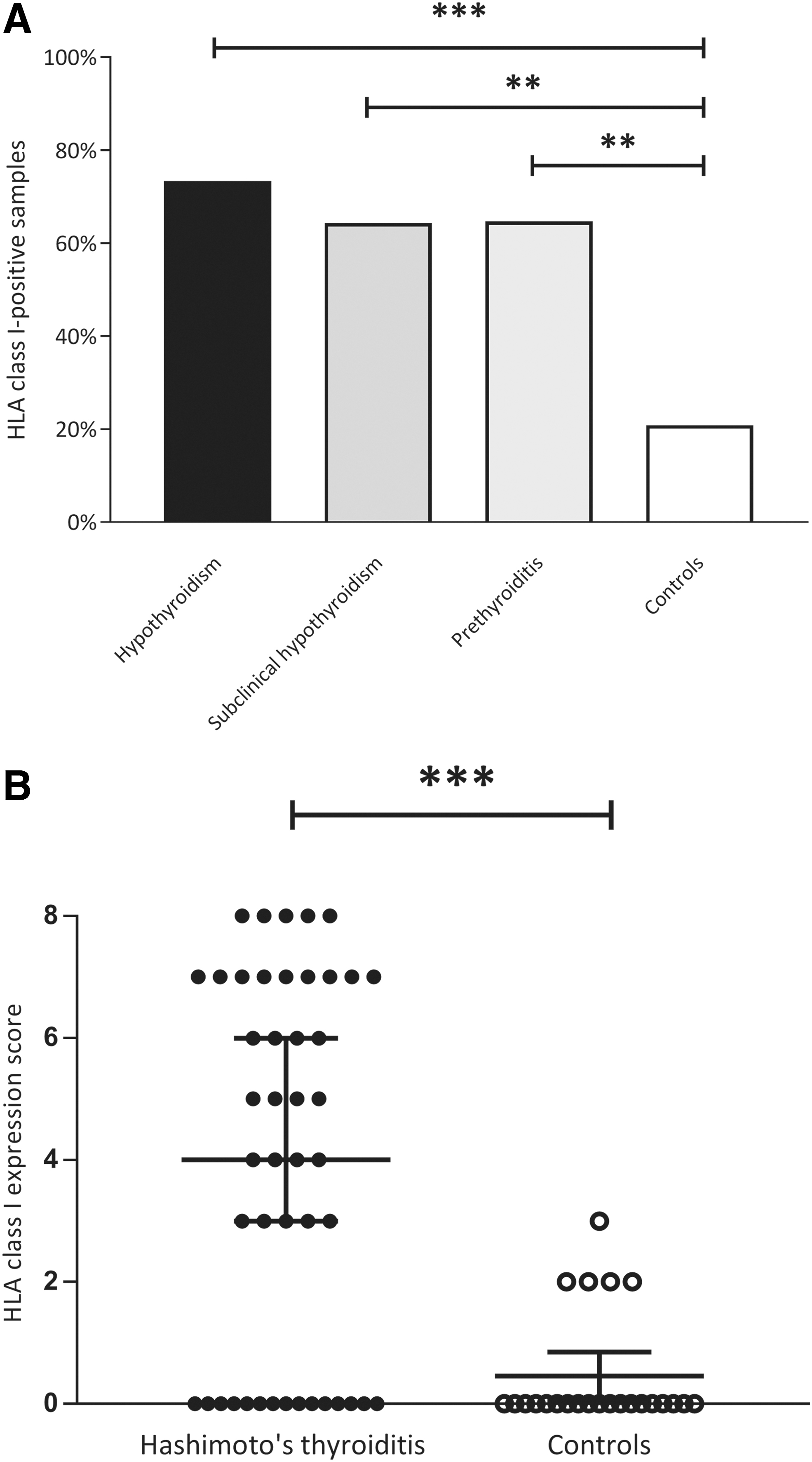
HLA class I immunodetection and HLA class I expression score. All three clinical subgroups showed a significantly higher number of HLA class I-positive samples compared with controls. (
The mean HLA class I expression score (Allred score) was 3.9 ± 3.1 in HT patients and 0.5 ± 0.9 in controls (p < 0.001) (Fig. 2B). Each subgroup had a significantly higher HLA class I expression score compared with the control group: 4.7 ± 3.3 in the subgroup with overt hypothyroidism, 3.0 ± 2.6 in the nontreated euthyroid thyroiditis group, and 4.1 ± 0.8 in the subclinical hypothyroidism group. There were no statistically significant differences in HLA class I expression score between the three subgroups.
When considering only the HLA class I-positive samples (HLA class I expression score ≥1), there was still a significant difference in HLA class I expression score between HT patients (5.7 ± 1.8) and controls (2.2 ± 0.4) (p < 0.001). Moreover, there were no samples with HLA class I expression score >3 in the control group. There was a significant positive correlation between TSH and HLA class I expression score (ρ = 0.458, p = 0.001) and a significant negative correlation between fT4 and HLA class I expression score (ρ = −0.379, p = 0.009). However, when using binary logistic regression TSH level and fT4 level did not influence HLA class I positivity.
TPO-Ab is the hallmark antibody in HT, and higher TPO-Ab titer levels have been associated with an increased likelihood of developing manifest hypothyroidism (37). There was a wide range of TPO-Ab levels within the HT group, ranging from 53 to 22,795 kIU/L (1320.7 ± 3330.0 kIU/L) (Table 1). TPO-Ab levels were higher (7708 ± 4012 kIU/L) in HLA class I-positive HT patients, than in HLA class I-negative HT patients (520 ± 356 kIU/L), however the difference did not reach statistical significance (p = 0.077) (Fig. 3). There was a significant correlation between TPO-Ab and HLA class I expression score (ρ = 0.358, p = 0.015). Assessed with binary logistic regression, TPO-Ab, TSH, fT4, and age did not significantly influence HLA class I positivity.

HLA class I and association with TPO-Ab TPO-Ab titers were higher in HLA class I-positive HT patients than in HLA class I-negative HT patients. However, the difference did not reach statistical significance. Bars represent median and 95% CI. NS, not significant; TPO-Ab, thyroid peroxidase antibodies.
Antiviral tissue response proteins
STAT1 and PKR are both antiviral proteins induced by interferons or viral products. The antiviral enzyme PKR was detected in three out of six HT samples and in one out of four control samples. In two of the cases, PKR was colocalized with VP1 (Fig. 4A and Table 2). STAT1 is a cytoplasmic protein, which upon activation relocates to the nucleus and initiates transcription of antiviral response factors. We analyzed STAT1 in a subset of samples (six HT and four controls). We found STAT1 in five out of six HT samples and in two out of four control samples (Table 2). STAT1 expression was colocalized with HLA class I (Fig. 4B), and both cytosolic and nuclear STAT1 expression was observed.
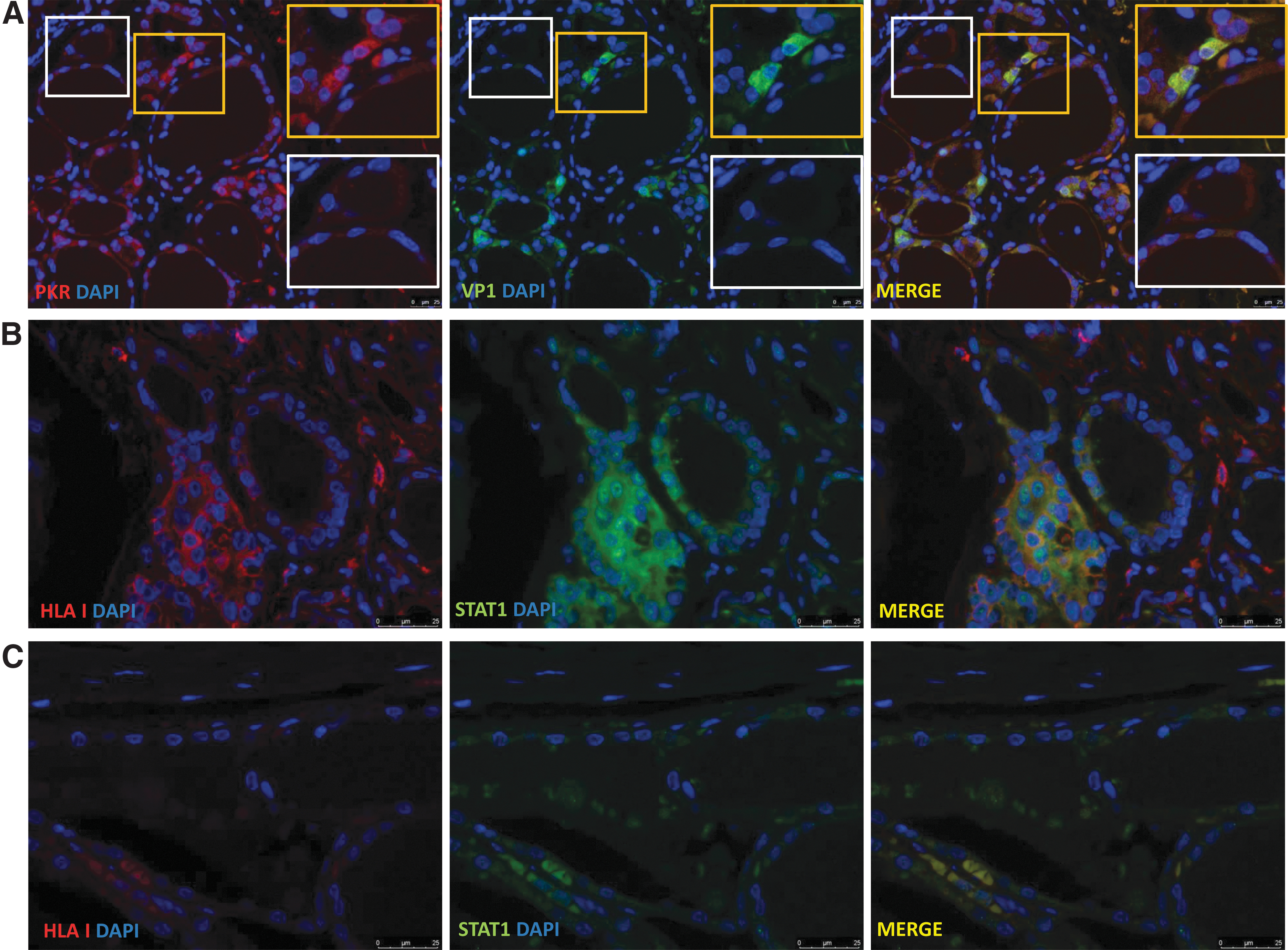
Combined immunofluorescence of PKR/VP1 and HLA class I/STAT1 (
Sequential Immunofluorescence in a Subset of the Samples
Ten samples were stained sequentially with immunofluorescence for HLA class I, STAT1, PKR, and VP1. The table shows that some samples were positive for HLA class I, in addition to the antiviral proteins STAT1 and PKR. A few samples were even positive for HLA class I, STAT1, PKR, and the viral capsid protein VP1.
HLA, human leukocyte antigen; PKR, protein kinase R; STAT1, signal transducer and activator of transcription 1; VP1, enteroviral capsid protein 1.
VP1 detection
We confirmed earlier findings indicating the presence of VP1 in HT, with no significant difference between patients and controls (10). However, there were more, although not significantly, VP1+ thyrocytes in the HT samples (20.1% ± 16.4%) than in the control samples (14.9% ± 10.5%) (Fig. 5A). VP1 immunoreactivity was found in 10 out of 46 samples (21.7%) in the HT group, and in 10 out of 24 samples (41.7%) in the control group. Eight of 46 (17.3%) HT samples had both HLA class I positivity and VP1 immunoreactivity, whereas only 2 out of 24 (8.3%) of the controls had both HLA class I and VP1 immunoreactivity (p = 0.304) (Fig. 5B). There was a significantly higher proportion of HT samples (23 out of 46, 50.0%) that were VP1-negative and HLA class I-positive, compared with controls (3 out of 24, 12.5%) (p = 0.002) (Fig. 5B). There was no significant correlation between HLA class I expression score and the number of VP1+ thyrocytes.
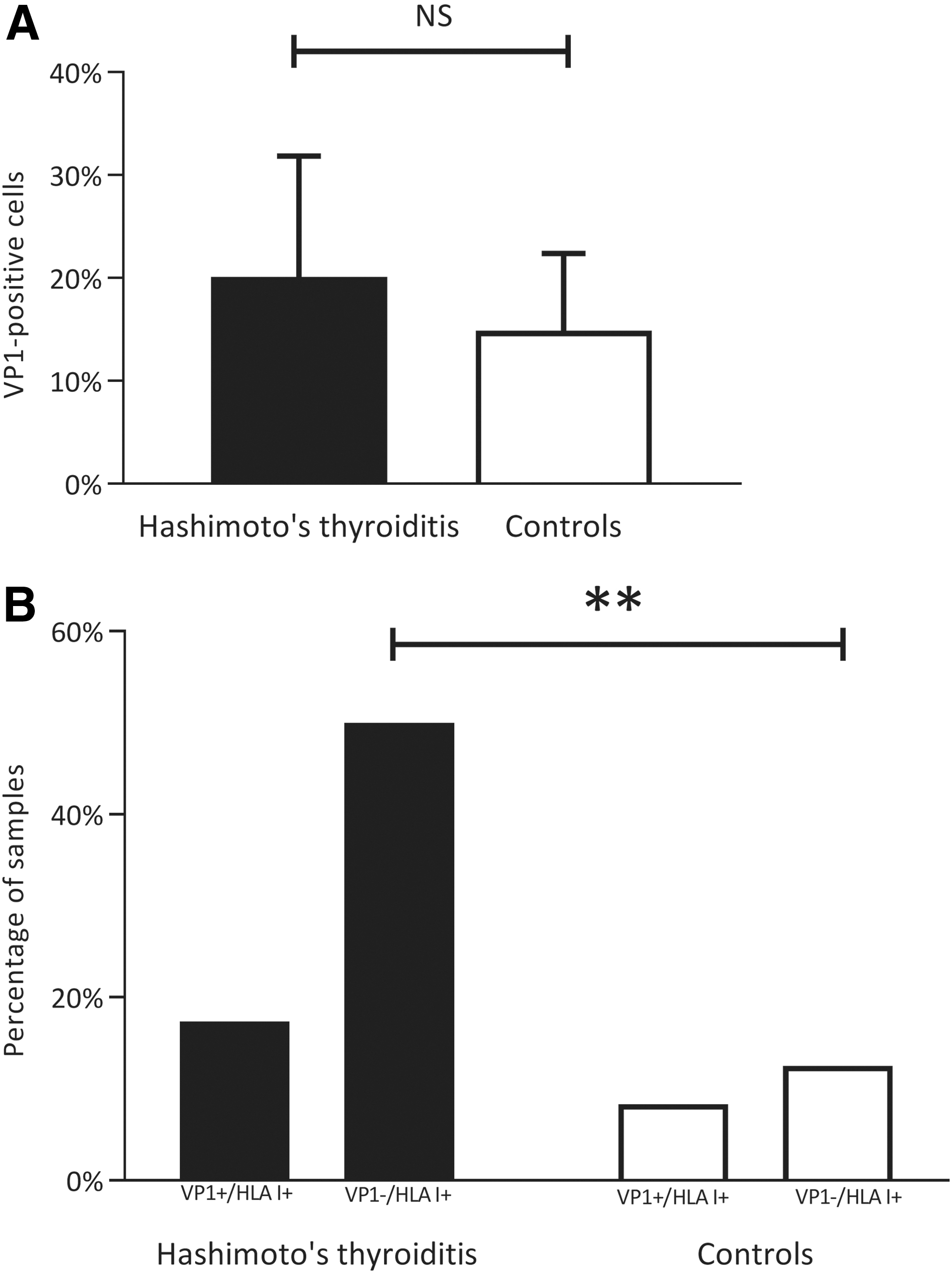
VP1 assessment in HT samples and controls. (
Myxovirus resistance protein and CD8+ T cells
We previously reported an increased expression of the type 1 interferon downstream response protein MxA and an increased density of CD8+ T cells in this HT cohort (13). When combining previous MxA results with the current HLA class I findings, we found a significant positive correlation between HLA class I score and number of MxA+ cells per mm2 thyroid tissue (ρ = 0.572, p = 0.001). Moreover, upon examination of HT samples only, we found significantly more MxA+ cells in the HLA class I-positive samples (112.8 ± 177.7) than in the HLA class I-negative cases (5.0 ± 5.7) (p = 0.030) (Fig. 6A). Additionally, there was a significant correlation (ρ = 0.605, p < 0.001) between HLA class I expression score and CD8+ T cells per mm2 . Moreover, CD8+ T cells were found in the HLA class I-positive HT samples, but not in the HLA class I-negative HT samples (Fig. 6B).
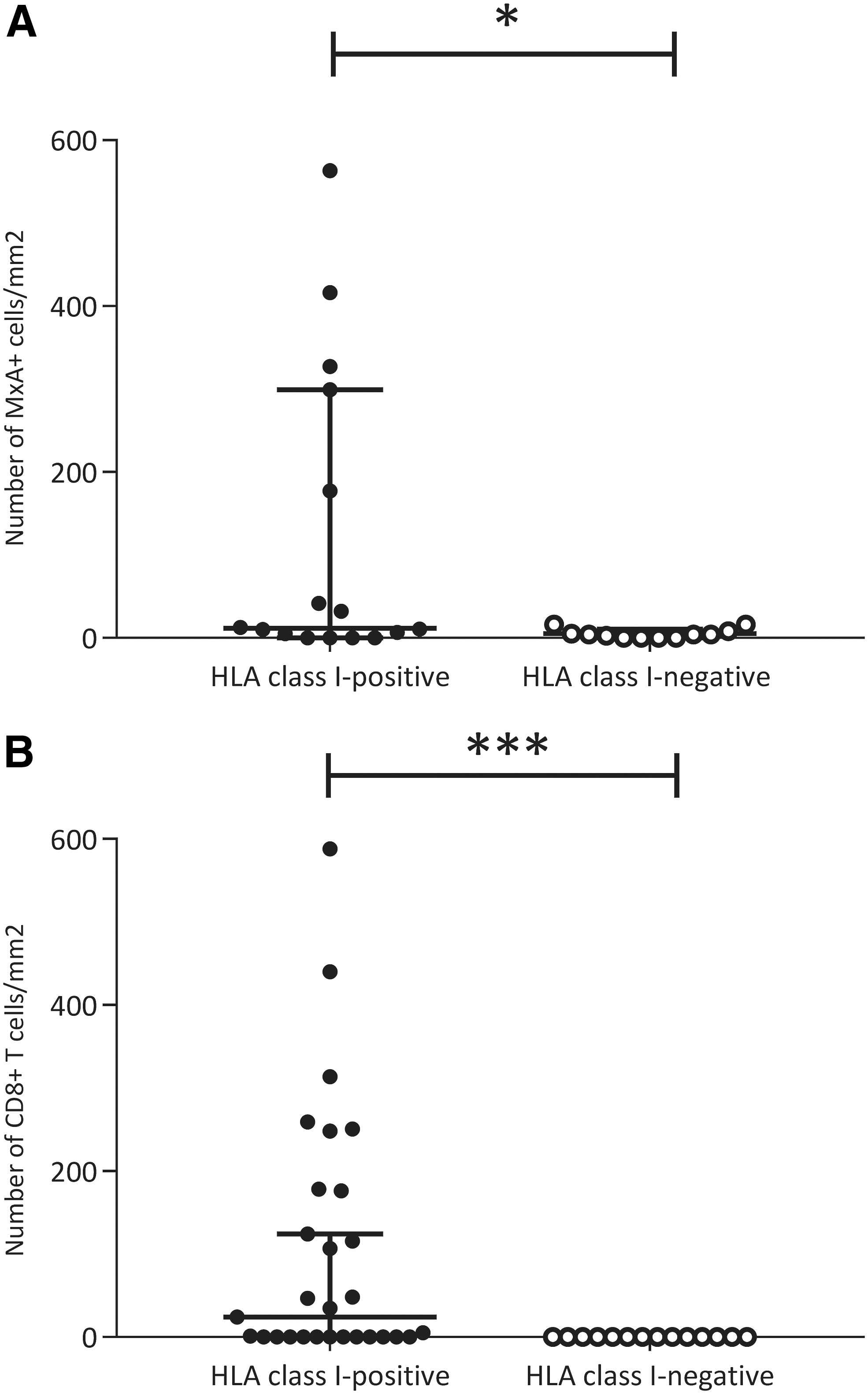
HLA class I positivity and associations with previously analyzed MxA and CD8+ T cells. (
Discussion
The main findings of the study presented here are that (i) the CAR-SIV isoform is present in thyrocytes; (ii) HLA class I is upregulated in thyrocytes of HT thyroid tissue; (iii) both STAT1 and PKR are present in thyrocytes from HT patients; and lastly, (iv) that VP1 is present in thyroid tissue from both HT patient samples and controls.
We observed an upregulation of HLA class I in all three clinical stages of HT, in a large collection of thyroid tissue samples from mainly newly diagnosed HT patients (Fig. 1B and Fig. 2A). Our results support previous findings of upregulated HLA class I in HT (38,39). There was a significant correlation between thyroid function and HLA class I expression; lower thyroid function levels were associated with higher HLA class I expression scores. However, TSH level and fT4 level did not influence HLA class I positivity.
Thyroid cell lines have previously been successfully infected by enterovirus (40). We detected a granular distribution of CAR-SIV in all investigated thyroid samples, thus confirming that thyroid cells are potentially susceptible to infection by enteroviruses (Fig. 1A). Interestingly, a granular pattern of the specific SIV-isoform of CAR detected in our samples was recently detected in abundance in pancreatic beta cells, where it was mainly associated with insulin secretory granules, suggesting a novel role for the receptor in granule maturation and trafficking (16). In thyroid tissue, it remains to be explored whether CAR-SIV serves this and/or other functions separate from mediating viral entry. VP1 was found in both HT patient samples and control samples, thus adding to the evidence of thyroid tissue being susceptible to enterovirus.
STAT1 expression was monitored using an antiserum, which detects both phosphorylated and nonphosphorylated forms and was present in both the cytosol and nucleus of thyrocytes, and its expression correlated with HLA class I (Fig. 4B). Viral infection induces production of type 1 interferons by plasmacytoid dendritic cells, and HLA class I is upregulated in response to interferon-α or interferon-γ in a manner, which can be dependent on STAT1 activation (41). Our results support these findings, as HLA class I was found in the same thyroid cells as STAT1 (noting that we were not able to distinguish between phosphorylated STAT1 and total STAT1). Moreover, both plasmacytoid dendritic cells and MxA were previously found in the same thyroid tissue samples as those used in this study (13). In addition, we demonstrate the presence of PKR, an antiviral enzyme that is known to interact with STAT1. Finally, we found colocalization of PKR and VP1, implying that enteroviral infection may lead to PKR induction (Fig. 4A).
With the methodology provided in this study, we did not find convincing evidence of current enterovirus infection in most of the samples. However, as previously reported by our group, tissue destruction, lymphocyte infiltration, and few remaining thyroid cells were found in HT thyroid specimens, even in early stages of the disease (13). Therefore, we cannot exclude that enterovirus infection occurred earlier.
We hypothesize that the increased HLA class I expression is due to an upregulated immune response following viral infection. MHC class I, Natural Killer (NK) cells, and interferons are all essential for antiviral defense. MHC class I presents viral fragments to CD8+ T cells, which upon activation, initiate cell destruction by releasing interferons. In contrast to CD8+ T cells, NK cells are not dependent on the MHC complex to identify and destroy infected cells. On the contrary, the “killing” action of NK cells is inhibited by self-MHC class I, because it activates an inhibitory receptor on NK cells. Thus, expression of MHC class I is fundamental for evading NK-mediated destruction. A number of viruses has developed immune escape strategies to establish themselves within the host. Several of these strategies are directed against antigen presentation by MHC class I. Some viruses such as cytomegalovirus, downregulate MHC class I to avoid CD8+ T cell-mediated attacks (18). However, common viruses such as hepatitis C virus, flavivirus, herpesvirus, rhinovirus, and hantavirus induce upregulation of MHC class I (19 –25).
Aberrant expression of HLA class II in AITD thyroid tissue has been reported, arguing that thyroid cells are complicit in inducing the autoimmune response (42). Moreover, several HLA class II genes are correlated with autoimmune susceptibility. On the other hand, the role of HLA class I in autoimmune diseases is less firmly established. Nevertheless, it has been shown that specific HLA class I alleles are associated with type 1 diabetes (43). As far as we know, this association has not been studied in autoimmune thyroiditis.
Viral infections have been implicated as triggers for various autoimmune diseases; type 1 diabetes being the most studied entity. Emerging evidence points to enteroviruses as being infective agents that can give rise to persistent infection in the pancreas, and ultimately lead to autoimmune destruction of beta cells (44 –48). Enterovirus, hepatitis C virus, herpesvirus, parvovirus, and many other viruses have all been associated with the development of AITD (5 –9,12). HT and type 1 diabetes share many common features; for example, active endocrine cells are destructed in both diseases, and the absence of the hormone normally produced by the destroyed cells, ultimately causes disease. Interestingly, HLA class I upregulation in islet cells from patients with type 1 diabetes has recently been confirmed at both the protein and RNA levels (49 –51). Five controls had an increased HLA class I expression, one of which had a preexisting autoimmune disease. However, the intensity and proportion of HLA class I staining was lower in the controls (HLA class I score <3) compared with HT patients. We cannot exclude that the five controls with increased HLA class I are at risk for developing autoimmune diseases.
We demonstrate not only increased HLA class I expression, but also the presence of the virus receptor CAR-SIV (Fig. 1A) and the viral response proteins STAT1 (Fig. 4B) and PKR (Fig. 4A) in thyroid tissue from HT individuals. Moreover, there was a strong correlation between previously found CD8+ T cells and HLA class I in this cohort (Fig. 6B). This suggests that the upregulated HLA class I found in our samples may present antigen to CD8+ T cells and thereby prompt an immunological response. Overall, this can be interpreted as part of the immune response to viral infection. Interestingly, a recent study showed that transfection of a thyroid cell line with dsRNA caused upregulation of HLA class I, PKR, STAT1, toll-like receptor 3, and other immune response proteins associated with viral infections (52), thus confirming that viral products can cause a response in thyroid cells similar to our findings. Nevertheless, there is still a lack of substantial evidence of viral presence in thyroid tissue.
We were not able to detect enterovirus with the limited methods used in this study. However, the infection might occur on such a modest scale that it largely escapes detection with antibody-based techniques. It is difficult to provide unequivocal proof of the presence of virus in tissue samples using indirect methods. In addition, extensive fibrosis, tissue destruction, and lymphoid displacement in thyroid tissue limit the possibility of finding virus. Our group previously reported the results for enterovirus RNA by in situ hybridization with similar results to that for VP1 (13). Finally, the timeline of disease progression in HT is difficult to establish (for example, some patients have high titers of TPO-Ab but never develop overt hypothyroidism). We speculate that viral infection might initiate the autoimmune process, but that the primary infection is cleared or undetectable by the time of clinically significant disease. A recent study found that patients with a known genetic predisposition for AITD have an increased expression of another AITD susceptibility gene when exposed to products of microbial infection (53). This interplay of genetic predisposition and infectious insults could explain why common infections give rise to autoimmunity in some but are negligible in others.
Our study has several limitations. Due to limited resources, we did not analyze PKR, STAT1, or CAR-SIV in all samples. Moreover, the size of the control group is limited.
In conclusion, we report the presence of the viral receptor CAR-SIV, in thyroid tissue. We also confirm that HLA class I upregulation is a significant feature of HT. In addition, we demonstrate that STAT1 is colocalized with HLA class I in thyrocytes and that PKR is colocalized with VP1, which is potentially indicative of an intracellular host antiviral response. Taken together with the previously reported increase in MxA, plasmacytoid dendritic cells and CD8+ T cells in the same cohort, our results support the hypothesis of an association between enteroviral infections and HT. Our study implies that antigen-presenting HLA class I elicits a CD8+ T response, and activates interferon downstream responses, such as MxA, PKR, and STAT1 induction in HT thyroid tissue. However, the pathogenesis of HT is complex and our knowledge of the numerous immunological pathways leading to lymphocytic displacement of thyroid cells is still limited. Hence, we call for further clarification of the pathogenesis and final confirmation of viral presence in HT.
Footnotes
Acknowledgments
The authors would like to thank Nina Gjerlaugsen at the Hormone Laboratory at Oslo University Hospital in Oslo, and Christine Flaxman and Marie Louise Zeissler at the Islet Biology laboratory at the University of Exeter for technical support and advice. Additionally, they are thankful to Professor Frode Jahnsen at the Department of Pathology and Center for Immune Regulation at Oslo University Hospital for technical microscopy support and useful comments on the article. Professor Kristian Folkvord Hanssen at the University of Oslo provided valuable comments on the article. Finally, they are grateful to all study participants and patients, without whom this work could not have been done.
Author Contributions
T.W. contributed to IHC examinations, data collection, analysis, and interpretation; and drafting of the article. S.S.H. contributed to all parts of the study; study design, clinical coordination, and patient recruitment; data collection, analysis, and interpretation; and drafting of the article. T.H.P. contributed to the surgery and writing of the article. S.J.R. and N.G.M., contributed to the IHC analysis, data analysis and interpretation and writing of the article. K.D.-J., as the principal investigator of the study, had the initial idea of the study and contributed to the study design; funding; regulatory issues; international collaboration; data collection, analysis, and interpretation; and writing of the article. T.W., S.S.H., and K.D.-J. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Author Disclosure Statement
The authors have nothing to disclose, and no competing financial interests exist.
Funding Information
South-Eastern Norway Regional Health Authority (HSØ grants to S.S.H and K.D.J.) financed this work. The faculty of Medicine at the University of Oslo provided a traveling grant. We are pleased to acknowledge financial support from the European Union's Seventh Framework Program PEVNET (FP7/2007–2013) (grant number 261441 to S.J.R. and N.G.M.). The participants of the PEVNET consortium are described at
Supplementary Material
Supplementary Tables S1
Supplementary Tables S2