
Abstract
Background:
Phosphorylation of the intracellular domain of the EPHA2 receptor tyrosine kinase (RTK) on serine 897 (S897) has been demonstrated to mediate EPHA2 oncogenic activity. Here, we show that in thyroid cancer cells harboring driver oncogenes that signal through the extracellular regulated kinase (ERK1/2) signaling pathway [rearranged RET RTK (RET/PTC), KRAS(G12R), or BRAFV600E oncogenes], EPHA2 is robustly phosphorylated on S897. EPHA2 S897 is embedded in a consensus sequence for phosphorylation by the AGC family kinases, including p90RSK (ribosomal protein S6 kinase), a direct ERK1/2 target.
Methods:
We show that recombinant p90RSK phosphorylates in vitro EPHA2 S897 and that treatment with chemical inhibitors targeting p90RSK or other components of the ERK1/2 pathway blunts S897 phosphorylation.
Results:
RNA interference-mediated knockdown combined with rescue experiments demonstrated that EPHA2 S897 phosphorylation mediates thyroid cancer cell proliferation and motility.
Conclusions:
These findings point to EPHA2 S897 as a crucial mediator of the oncogenic activity of the ERK1/2 signaling cascade in thyroid cancer.
Introduction
Thyroid carcinomas, which originate from epithelial follicular cells, are subclassified into differentiated thyroid carcinoma (DTC), papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma, poorly differentiated thyroid carcinoma, and anaplastic thyroid carcinoma (ATC). PTC accounts for >80% of thyroid carcinoma cases (1 –4).
Oncogenic conversion of the extracellular regulated kinase (ERK1/2) signaling cascade is the most important driver for PTC formation. Follicular-variant PTCs commonly have mutations in RAS (or less commonly the BRAFK601E mutation) and classic-variant PTCs are typically associated with BRAFV600E (or other less common BRAF mutations) mutation or rearrangements of receptor tyrosine kinases (RTKs), including RET (RET/PTC oncogenes) and NTRK family members (5 –7). Oncogenic activation of the PI3K-AKT-mTOR pathway frequently cooperates with the ERK1/2, particularly in most aggressive thyroid carcinoma subtypes (8 –10).
Through a loss-of-function siRNA-mediated screening, we have recently identified EPHA2 as an essential component of the thyroid cancer cell kinome involved in thyroid cancer cell viability (11). Erythropoietin-producing human hepatocellular carcinoma (EPH) represents the largest subfamily of RTKs (12,13). EPH RTKs play a critical role in different processes, including axon guidance, synaptic plasticity, vascular development, tissue-border formation, and cell migration (12,13).
EPHA2 upregulation, often accompanied by the loss of its cognate ephrin ligand, has been documented in several human malignancies, including breast, lung, prostate, skin, esophageal, gastric, and renal carcinomas (14 –24) as well as in thyroid carcinoma (25,26). In cancer cells, EPHA2 commonly functions downstream of growth factor receptors and independently of ephrin stimulation (13). In turn, EPHA2 oncogenic effects can be mediated by phosphorylation on serine 897 (S897) that maps in the EPHA2 cytosolic domain between the C-terminal domain of the kinase and the sterile α motif (SAM) domain (27).
Miao et al. demonstrated that S897 EPHA2 phosphorylation mediated by AKT is crucial for glioma cell invasive phenotype and stem cell properties (28,29). More recently, Zhou et al. reported that inflammatory cytokines are able to promote phosphorylation of EPHA2 at S897 mediated by the RSK and this fosters breast cancer cell metastatic properties (30).
Here we show the role of RSK-dependent EPHA2 phosphorylation at S897 downstream of the ERK1/2 pathway in RET- and BRAF-mediated thyroid tumorigenesis.
Materials and Methods
Cell cultures
TPC1 and BCPAP PTC cell lines are positive for CCDC6-RET (RET/PTC1) rearrangement or BRAFV600E point mutation, respectively. 8505C, OCUT-1, and SW1736 ATC cell lines are positive for BRAFV600E mutation; and CAL62 ATC cells are positive for KRAS G12R mutation (31). Nthy-ori 3-1 (hereafter referred to as NTHY) is a human follicular epithelial cell line, derived from normal thyroid, immortalized by the SV40 large T gene. TPC1 cells were originally obtained by M. Nagao (Carcinogenesis Division, National Cancer Center Research Institute, Tokyo, Japan). BCPAP were obtained from the primary source (N. Fabien, CNRS URA 1454, University of Medecine Lyon-Sud, Oullins, France).
8505C and CAL62 anaplastic carcinoma cells were purchased from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany). Nthy-ori 3-1 was purchased from the European Collection of Authenticated Cell Cultures (ECACC, Wiltshire, United Kingdom). OCUT-1 a gift from K. Hirakawa and N. Onoda (Department of Surgical Oncology, Osaka City University Graduate School of Medicine, Osaka, Japan) and SW1736 cells were a gift from C.H. Heldin (Ludwig Institute for Cancer Research, Uppsala University, Uppsala, Sweden).
NTHY and 8505C cells were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM
PC Cl 3 (hereafter referred to as PC) is a differentiated nontumoral thyroid follicular cell line derived from 18-month-old Fischer rats. PC cells were cultured in Coon's modified Ham F12 medium (LONZA, Walkersville, USA) supplemented with 5% calf serum (CS) (LONZA) and a mixture of six hormones, including thyrotropin (10 mU/mL), hydrocortisone (10 nM), insulin (10 μg/mL), apo-transferrin (5 μg/mL), somatostatin (10 ng/mL), and glycyl-histidyl-lysine (10 ng/mL) (Sigma-Aldrich Chemie GmbH, Steinheim, DE). Stably transfected PC RET/PTC1 and BRAFV600E
cells as well as doxycycline-inducible RET/PTC3 cells (a kind gift from Dr. J.A. Fagin) have been described previously (32
–34). Phoenix human embryonic kidney cells (hereafter referred to as φχ) were grown in DMEM supplemented with 10% FBS, 2 mM
Chemical treatments were performed in low serum conditions (2.5% FBS or CS) for the indicated time points. Chemical inhibitors used were as follows: PLX4032 (vemurafenib), U0126 (Cell Signaling Technology, Danvers, MA), BI-D1870 (Selleckchem, Houston, TX), wortmannin (Cell Signaling Technology), and doxycycline (1 μg/mL; Sigma-Aldrich, Inc., Saint Louis, MI).
Cell proliferation assay and foci assay
A total of 5 × 104 TPC1 and 8505C or 8 × 105 PC-PTC1 and PC-BRAFV600E cells were plated in 60-mm dishes. Cells were counted at 48 or 72 hours after plating. Each count was performed in triplicate and each point was the mean value of three dishes. Foci formation assay was performed in 8505C cells transfected with shSCR and shEPHA2, cultured for 2 weeks in the presence of 5% serum, fixed with 10% (v/v) methanol for 15 minutes, and stained with crystal violet for 30 minutes for colony visualization and counting. Each count was performed in triplicate.
Wound healing assay
8505C and TPC1 cells were grown for 60 hours to form monolayers. A wound of ∼300 μm width was inflicted with a sterile pipette tip. The culture medium was refreshed to remove nonadherent cells. Wound closure (healing) was recorded by microphotographs taken with the Leica DM IL light microscope (Leica Microsystems, Wetzlar, Germany) immediately and after 24 or 48 hours. Size of the wound gap was measured and migration was expressed as percentage of wound closure, where 100% is the value obtained at 48 hours for control cells.
Matrigel assay
In vitro invasiveness through Matrigel was assayed using transwell cell culture chambers. Briefly, confluent cell monolayers were harvested with trypsin/EDTA and centrifuged at 800 g for 10 minutes. The cell suspension (2 × 105 cells/well) was added to the upper chamber of transwells on prehydrated polycarbonate membrane filter of 8 mM pore size (Costar) coated with 35 μg Matrigel (Collaborative Research, Inc.). The lower chamber was filled with complete medium. Cell dishes were incubated at 37°C in 5% CO2 and 95% air for 48 hours. Nonmigrating cells on the upper side of the filter were wiped off, and migrating cells on the reverse side of the filter were stained with 0.1% crystal violet in 20% methanol for 15 minutes, counted, and photographed. Pixel densities were measured and expressed as percentage of Matrigel invasion, where 100% is the value obtained for shSCR control cells.
Growth in 3D Matrigel
For outgrowth in Matrigel, 10,000 cells were mixed with 160 μL of Matrigel (BD Biosciences, San Jose, CA) and plated in 35-mm dishes containing a glass coverslip. After hardening, cells were overlaid with 2 mL growth medium and incubated at 37°C for up to 10 days.
Plasmid construction and site-directed mutagenesis
The plasmid encoding human green fluorescent protein (GFP)-tagged EPHA2 (RG205725) (GenBank accession number NM_004431) was purchased from Origene Technologies (Rockville, MD). GFP-EPHA2 was used as a template to generate the EPHA2 Ser897Ala (S897A) mutant by using the QuikChange Site-Direct mutagenesis kit (Stratagene/Agilent Technologies, Santa Clara, CA). Mutagenesis primers were designed according to the manufacturer's instruction and the Operon tool, and synthesized by the Ceinge Core Service Unit (Naples, IT). The mutation was confirmed by DNA sequencing performed by the Ceinge Core Service Unit. The pRK7-myr-RSK1 (8997) (GenBank accession number NM_001006665.1) plasmid was purchased from Addgene (Addgene, Cambridge, MA). Myr-AKT (activated AKT by the addition of a myristoylation site at the N-ter) and HA-MEKEE (activated MEK1 generated by replacing Ser-218 and Ser-222 by glutamic acid) plasmids were kind gifts of Dr. J.S. Gutkind (University of California and Moores Cancer Center, San Diego, CA).
RNA extraction and reverse transcription-PCR
Total RNA was isolated by the RNeasy Kit (Qiagen, Crawley, West Sussex, United Kingdom) and subjected to on-column DNase digestion with the RNase-free DNase set (Qiagen) following the manufacturer's instructions. The quality of RNA was verified by electrophoresis using 1% agarose gel and visualization with ethidium bromide. Random-primed first-strand cDNA was synthesized in a 50 μL reaction volume starting from 2 μg RNA by using the Gene Amp RNA PCR Core Kit (Applied Biosystems, Warrington, United Kingdom). PCR amplification was performed using the GeneAmp RNA PCR Core Kit system following the manufacturer's instructions. To exclude DNA contamination, each PCR was also performed on untranscribed RNA. Levels of GAPDH transcripts were used as a control for equal RNA loading. Reverse transcription-PCR products were loaded on 2% agarose gel, stained with ethidium bromide, and the image saved by the Typhoon 8600 laser scanning system (Amersham Pharmacia Biotech, Bucks, England).
Cell transfections
φχ cells were plated at 50% confluency in 60-mm poly-
EPHA2-specific shRNA expression vector and a scrambled control vector were purchased from OriGene Technologies. Experimental procedures were done as follows: the day before transfection, 8505C cells were plated in 35-mm dishes at 40% confluence in DMEM supplemented with 10% FBS without antibiotics. Two micrograms of sh-EPHA2 and control vector was transfected using the FuGENE HD transfection (Promega, Madison, WI) reagent according to the manufacturer's instructions. Seventy-two hours after transfection, the culture medium was supplemented with puromycin (Sigma-Aldrich) at a final concentration of 1 μg/μL for 14 days. Stably transfected cells were screened by RNA and protein expression.
8505C and PC cells were transfected by using the FuGENE reagent (Promega Co.). RSK knockdown experiments were performed with the ON-TARGETplus SMARTpool RSK1 (L-003025-00) and RSK2 (L-003026-00) human siRNAs provided by DharmaconGE (Milan, IT).
EPHA2 transient silencing was performed with the ON-TARGETplus SMARTpool EPHA2 (E-003116-00) (DharmaconGE). As suggested in the manufacturer's instructions, 10 μL of 5 μM siRNA was mixed with 2.5 μL of Dharmafect1 transfection reagent (Carlo Erba) in the Opti-MEM medium. The transfection mix was added to the cells and incubated for 48 hours.
In the rescue experiments, EPHA2 was knocked down in 8505C cells with a human-specific siRNA (SI02223508) from Qiagen, and in PC rat cells with a rat ON-TARGETplus SMARTpool (L-099402-02) provided by DharmaconGE. 8505C cells were transfected using 6.6 μL of 20 μM siRNA mixed with 200 μL of Opti-MEM medium and 13.2 μL of HiPerFect Transfection reagent (Qiagen) for 10 minutes. The transfection reaction was then applied to each plate.
EPHA2 knockdown in PC cells was performed by using the same conditions as described for RSK silencing. As negative controls for silencing experiments, AllStars Negative Control siRNA (SI03650318; Qiagen) or siGENOME Nontargeting siRNA (D-001206-13; Carlo Erba) were used. Following 36 hours of silencing, rescue was obtained by transient transfection of EPHA2 wt (4 μg) or EPHA2 S897A mutant (7 μg) with 12 μL of FuGENE (Promega Co.). Trypan blue exclusion test and dosage of cleaved-PARP in protein lysates were applied to exclude nonspecific toxicity of the transfection procedure (data not shown).
Protein experiments
Cell culture plates were washed two times with ice-cold PBS; cells were scraped in fresh lysis buffer containing 50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES; pH 7.5), 1% (vol/vol) Triton X-100, 150 mM NaCl, 5 mM EGTA, 50 mM NaF, 20 mM sodium pyrophosphate, 1 mM sodium vanadate, 2 mM phenylmethylsulfonyl fluoride (PMSF), and 1 μg/mL aprotinin. Lysates were clarified by centrifugation at 12,000 × g for 20–30 minutes and kept at 4°C during all passages. Protein concentration was measured by using a modified Bradford Assay (Bio-Rad Laboratories, Munich, Germany). Protein lysates were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Whatman Protran nitrocellulose membranes (PerkinElmer Health Sciences, Groningen, Netherlands).
For EPHA2 pull-down, after precleaning by incubation with gamma-bind G Sepharose beads (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), 500 μg of cell lysates in 500 μL volume was incubated overnight at 4°C (with gentle rotation) with 0.1 μg of recombinant Human EPHrin-A1 Fc protein (R&D Systems, Minneapolis, MN) for 1 mg of cell lysate. Ligand/receptor complexes were pulled down using 50 μL of protein-G sepharose beads for 1 hour at 4°C. The samples were centrifuged and washed in protein lysis buffer, eluted in sample buffer loading dye, boiled for 5 minutes, and run on 7.5% SDS-PAGE.
Immunoprecipitations were performed according to standard procedures. After blotting, membranes were incubated in 5% nonfat dry milk (Bio-Rad Laboratories) or TBS-BSA 5% (Sigma-Aldrich, Inc.) blocking solution for 1 hour at room temperature, and then with primary antibodies overnight at 4°C. After washes, the membranes were incubated for 1 hour at room temperature with HRP-conjugated anti-mouse or anti-rabbit secondary antibodies (dilution 1:3000) from Bio-Rad Laboratories, or with HRP-conjugated donkey anti-goat secondary antibodies (dilution 1:3000) from Santa Cruz Biotechnology (Heidelberg, Germany). Immunocomplexes were detected using the enhanced chemiluminescence (ECL) kit from Thermo Fisher Scientific (Rockford, IL); images were scanned with Epson Perfection V750 PRO and signal intensity was acquired by Cawomat 2000 IR.
Primary antibodies were directed against the following: Eck/EPHA2 clone D7 (05–480) (1:1000) from Millipore (Merck Millipore Corporation/Life Science, Darmstadt, Germany), phospho-Tyr1062 RET (AF5009) from R&D Systems (Minneapolis, MN), phospho-Ser897 EPHA2 D9A1 (6347), phospho-AKT Ser473 (9271), phospho-MEK1/2 Ser217/221 (9121), phospho-p44/42 MAPK (ERK1/2) Thr202/Tyr204 (4370), phospho-p90RSK Ser380 (9341), phospho-YB1 Ser102 C34A2 (2900), phospho-AKT substrate RXXS*/T* 110B7E (9614), phospho-AKT substrate RXRXXS*/T* 23C8D2 (10001), phospho-PRAS40 Thr246 (2640), AKT (9272), MEK1/2 (9122), p44/42 MAPK (9102), and RSK1/RSK2/RSK3 32D7 (9355) (Cell Signaling Technology). Antibodies for RSK1 (C-21), RSK2 (C-19), c-Myc 9E10 (sc-40), and actin (sc-1616) were from Santa Cruz Biotechnology. Antibody for BRAF (07–453) was from Upstate (Lake Placid, NY). Anti-RET is a polyclonal antibody raised against the tyrosine kinase protein fragment of human RET. Monoclonal anti-α-tubulin (T 9026) was from Sigma-Aldrich.
Dephosphorylation and phosphorylation protocols
φχ cells transfected with EPHA2 were lysed without phosphatase inhibitors in EDTA-free JS buffer according to the published procedure by Abcam. Thirty micrograms of crude extracts was treated with 1 U/μg of CIP (calf intestinal alkaline phosphatase; New England Biolabs, Beverly, MA) for 30 minutes at 37°C. The reaction was stopped with loading dye; samples were heated at 99°C for 5 minutes before SDS-PAGE. For dephosphorylation of nitrocellulose membranes, the membrane was blocked with 5% BSA in TBS with 0.1% of Triton X-100 for 1 hour at room temperature and then incubated with TBS-1% Triton with or without CIP (68 mU/μg) for 30 minutes in a 37°C water bath. Membranes were then incubated for one hour with the phospho-S897 EPHA2 antibody and subjected to ECL.
For recombinant protein kinase assay, 200 ng of recombinant full-length active RSK1 from SignalChem (R15–10G) was incubated with 20 ng of EPHA2 GST-tagged cytosolic domain from Life technologies (PV3688), as a substrate, in kinase buffer (25 mM MOPS [pH 7.2], 12.5 mM β-glycerophosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM DTT, and 50 μM ATP). Following incubation at 30°C for 15 minutes, the reaction was stopped by heating to 99°C in loading dye for 5 minutes.
For peptide kinase assay, EPHA2 peptides (WT:RVSIRLPSTSGSE, S897A:RVSIRLP
Xenografts in nude mice
Mice were housed in barrier facilities and 12-hour light/dark cycles and received food and water ad libitum at the Dipartimento di Medicina Molecolare e Biotecnologie Mediche (University of Naples “Federico II,” Naples, Italy). This study was conducted in accordance with the regulations of Italian Ministry of Health (approval number: 157/2015-PR) for experimentation on animals. All manipulations were performed while mice were under isoflurane gas anesthesia. No mouse showed signs of wasting or other signs of toxicity. 8505C cells transfected with scrambled or EPHA2 shRNA (10 × 106 per mouse) were inoculated subcutaneously into the right dorsal portion of 4-week-old male BALB/c nu/nu mice (The Jackson Laboratory). Tumor diameters were measured at regular intervals with calipers. Tumor volumes (V) were calculated with the following formula: V = A × B 2/2 (A = axial diameter; B = rotational diameter). Tumors were excised and fixed overnight in neutral buffered formalin and processed by routine methods.
Statistical analyses
Statistical analyses were performed using a paired, two-tailed Student's t test (GraphPad Prism 3.0; GraphPad Software, San Diego, CA), and differences were considered to be statistically significant at a value of p ≤ 0.05.
Results
EPHA2 S897 phosphorylation is upregulated in thyroid cancer cells
Western blot analysis of a panel of human thyroid cancer cell lines with endogenous levels of ERK1/2-driving oncoproteins, CCDC6-RET fusion (RET/PTC1) [TPC1 cells], KRAS G12R [CAL62 cells], and BRAFV600E (BCPAP, 8505C, SW1736, and OCUT-1 cells), demonstrated robust EPHA2 expression and phosphorylation on serine 897 (S897) compared with SV40-LT immortalized NTHY cells (Fig. 1A). Most of the cancer cell lines expressed phosphorylated MEK1/2, ERK1/2 (35) and p90RSK, a direct ERK1/2 effector through its phosphorylation on S380. In support of the specificity of the phospho-S897 EPHA2 antibody, CIP treatment of Phoenix cells transiently transfected with EPHA2 strongly reduced pS897 signal (Supplementary Fig. S2A) and S897 to Ala substitution in EPHA2 also reduced phospho-S897 antibody reactivity (Supplementary Fig. S2B).
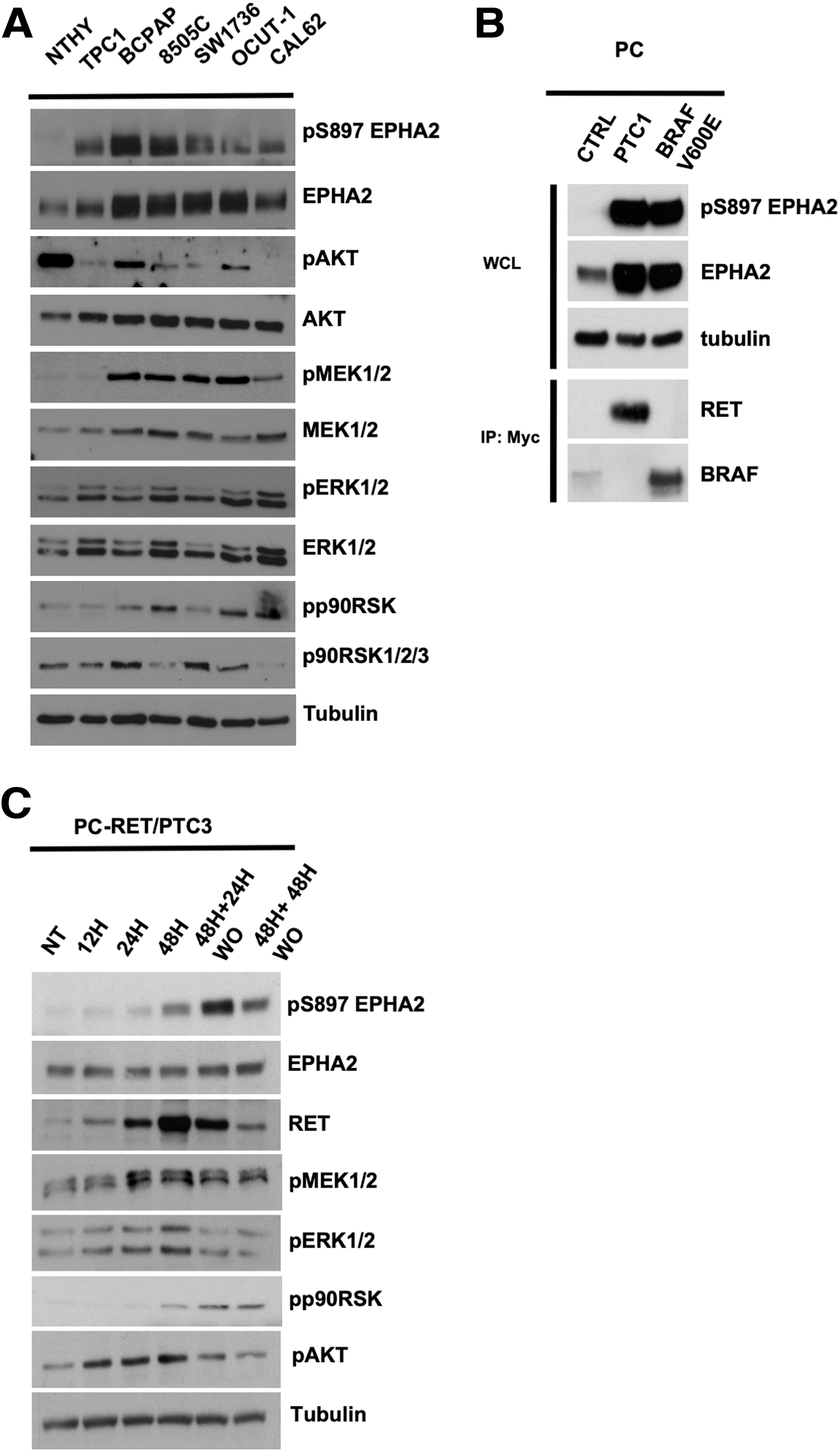
Expression and S897 phosphorylation of EPHA2 in thyroid cancer cells. (
To expand these findings, we used rat thyroid PC cells engineered to express RET/PTC1 or BRAFV600E. In these cells, EPHA2 expression was increased and phosphorylated on S897 compared with parental cells (Fig. 1B). The increased expression of EPHA2 in PC-RET/PTC1 or -BRAFV600E cells is consistent with the reported ability of ERK1/2 to induce EPHA2 gene transcription (36).
To dissociate EPHA2 phosphorylation from its increased expression, a short-term experiment was performed by using PC cells, in which expression of another RET/PTC variant (RET/PTC3, NCOA4-RET) was conditionally induced by doxycycline (32). RET/PTC3 upregulation resulted in a gradual and time-dependent increase of S897 phosphorylation, whereas EPHA2 expression levels did not change significantly at these time points. In parallel, RET/PTC3 induced an early (at 12–48 hours) activation of MEK1/2/ERK1/2 and AKT, and a delayed (at 48 hours) activation of p90RSK (Fig. 1C). Doxycycline washout was followed by a prompt decrease of phosphorylation of MEK1/2/ERK1/2 and AKT, while S897 and p90RSK phosphorylation persisted for at least 48 hours upon washout (Fig. 1C).
Altogether, these data proved robust EPHA2 phosphorylation on the activatory S897 residue in thyroid cancer cells harboring ERK1/2 -stimulating oncoproteins.
ERK1/2 and p90RSK inhibition attenuates EPHA2 S897 phosphorylation in thyroid cancer cells
To dissect the signaling cascade involved in EPHA2 S897 phosphorylation, RET/PTC1-positive TPC1 cells and BRAFV600E -positive 8505C cells were treated with chemical inhibitors of PI3K (wortmannin), MEK1/2 (UO126), or p90RSK (BI-D1870). Interestingly, in RET/PTC1-positive TPC1 cells, EPHA2 S897 phosphorylation responded to both MEK1/2/RSK and PI3K inhibition, suggesting that both pathways act on EPHA2 activation. The efficacy of drug treatments was confirmed by downregulated phosphorylation of MEK1/2/ERK1/2, AKT, as well as phosphorylation of RSK and its bona fide substrate YB1 (Fig. 2A, left panels). In BRAFV600E -positive 8505C cells, inhibition of BRAF and MEK1/2/RSK, but not PI3K blockade, attenuated EPHA2 S897 phosphorylation, suggesting that only the ERK1/2 pathway is involved in EPHA2 activation downstream of the oncogenic BRAF kinase (Fig. 2A, right panels).
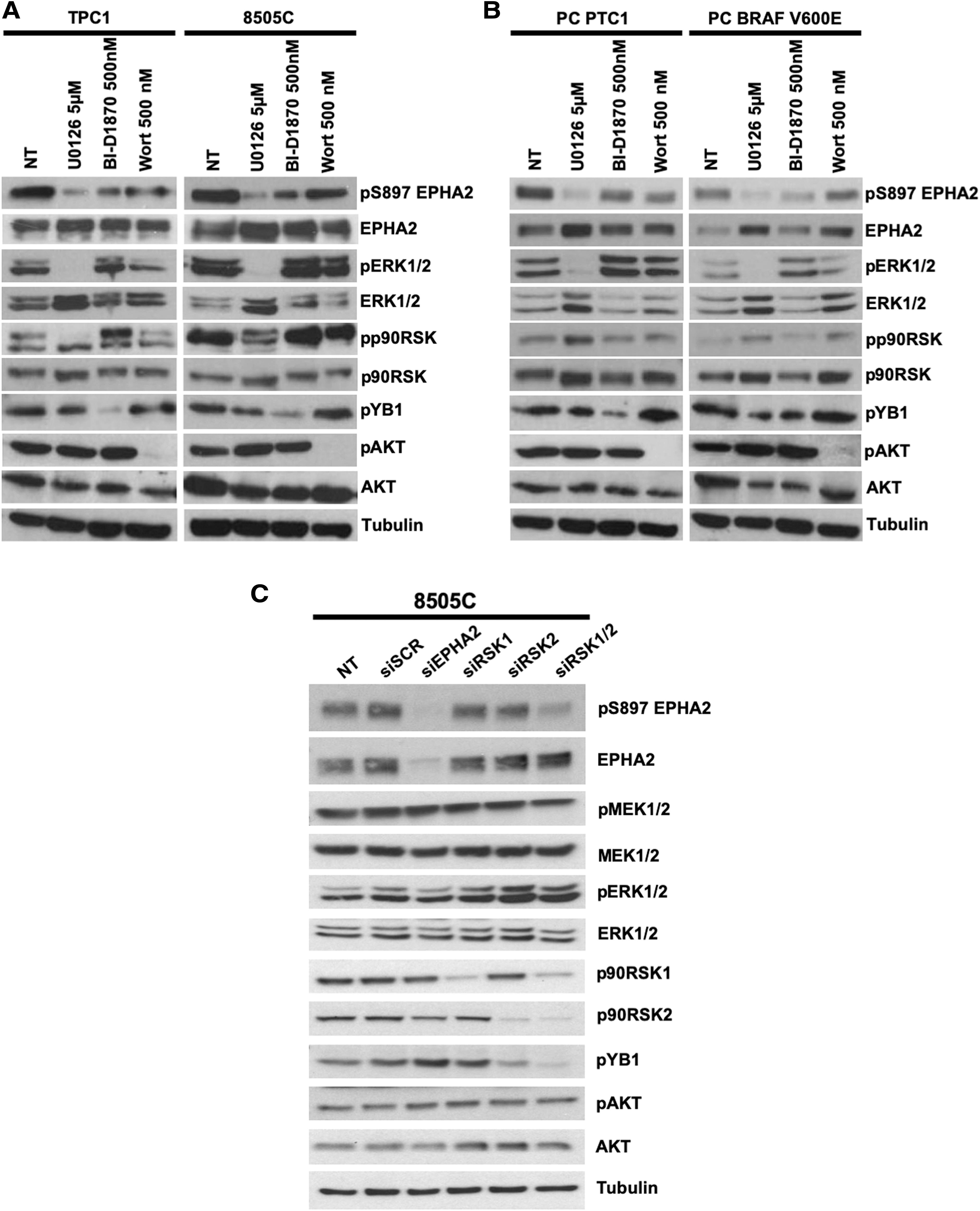
Modulation of S897 EPHA2 phosphorylation in thyroid cell lines with chemical inhibitors. Protein lysates (15 μg) were harvested from TPC1 or 8505C cells (
Similar results were obtained in PC cells ectopically expressing RET/PTC1 or BRAFV600E oncoproteins. In these cells, the inhibition of MEK1/2/RSK was effective at reducing pS897 EPHA2. Again, wortmannin had a modest effect in RET/PTC1 and a negligible one in BRAFV600E mutant cells, despite efficiently inhibiting AKT (Fig. 2B).
In addition, thyroid cancer cell lines harboring BRAFV600E (BCPAP and SW1736) or KRAS G12R (CAL62), BRAF inhibition with PLX4032 (only in BRAF mutant), MEK1/2 inhibition with U0126 (in all of them), or p90RSK inhibition with BI-D1870 were effective at inhibiting pS897 EPHA2, while PI3K-AKT inhibition with wortmannin had only minor effects (Supplementary Fig. S3).
Out of four RSK family members (RSK1–4), RSK1 and RSK2 were expressed in all tested cell lines at the mRNA level (Supplementary Fig. S4). Thus, siRNA-mediated RSK1/2 double knockdown significantly reduced phosphorylation of S897 EPHA2, as well as of the RSK substrate YB1, while knockdown of either RSK1 or RSK2 alone was not sufficient to significantly affect both phosphorylations (Fig. 2C). Total and phosphorylated MEK1/2 and AKT levels did not change significantly. EPHA2 silencing was used as a control.
Altogether, these data show that ERK1/2 signaling through p90RSK is able to sustain S897 EPHA2 phosphorylation in the BRAFV600E mutant and, in adjunct to AKT, in RET mutant thyroid cancer cells.
p90RSK is an EPHA2 S897 kinase
EPHA2 S897 is embedded in the consensus phosphorylation sequence (RLPS897 single letter code), featuring an arginine residue at position-3 relative to the phosphorylated site, for kinases of the AGC (PKA, PKG, PKC) family (37). Accordingly, the EPHA2 protein, purified by pull-down of thyroid cancer cell lysates with a recombinant ephrinA1 ligand, was recognized by a phospho-specific antibody targeting the sequence RXXpS/T (containing an arginine at position-3), but not by an antibody targeting the sequence RXRXXpS/T (requiring also an arginine at position-5 that is lacking in EPHA2) used as a control (Supplementary Fig. S5).
It has been previously reported that EphA2 S897 phosphorylation can be mediated by different AGC kinases, such as AKT (28,29), p90 ribosomal S6 (RSK) kinase (p90RSK) (30), and PKA (38). We reasoned that p90RSK, as a direct downstream effector of the ERK1/2, was a candidate kinase for the phosphorylation of EPHA2 S897 in thyroid cancer cells. Thus, to confirm that p90RSK was able to mediate EPHA2 S897 phosphorylation, we transfected φχ cells with active forms of AKT (Myr-AKT), MEK1/2 (HA-tagged MEKEE), or RSK1 (Myr-RSK1); full-length GFP-tagged EPHA2 and GFP alone were also used. In this setting, Myr-RSK1 was a stronger inducer of S897 phosphorylation than AKT or MEK1/2 (Fig. 3A).

p90RSK mediates EPHA2 S897 phosphorylation. (
Thus, we performed an in vitro kinase assay using full-length GST-tagged recombinant p90RSK1 as a kinase and cytoplasmic recombinant EPHA2 (cEPHA2) protein as a substrate. Figure 3B shows that when incubated alone cEPHA2 already featured detectable S897 phosphorylation, which was abrogated upon treatment with CIP. Incubation with p90RSK1 strongly increased S897 phosphorylation levels and this was almost completely erased by CIP (Fig. 3B).
Finally, to map the p90RSK1 phosphorylation target sequence, we synthesized an EPHA2 peptide spanning S897 (18) or mutated versions, in which either all the 4 serine/threonine residues (4S/T→A) or only S897 (S897A) was replaced by alanine. As a further control, we also used a peptide in which the RXXpS consensus was disrupted by replacing the arginine at position-3 with a lysine (R894K). Incubation with recombinant p90RSK1 and γ-32P ATP induced robust incorporation of 32P in the wild-type peptide but not in its mutated versions, indicating that the S897 consensus sequence is a direct substrate of p90RSK1 in vitro (Fig. 3C).
Altogether, these findings demonstrate that p90RSK is able to phosphorylate S897 both in intact cells and in the test tube.
S897 phosphorylation of EPHA2 promotes growth and motility of thyroid cancer cells
SiRNA-mediated EPHA2 knockdown reduced the number of 8505C cells compared with control siRNA (Fig. 4A). As a control, cotransfection with a GFP-tagged EPHA2 was able to partially rescue the number of cells. Importantly, a nonphosphorylatable S897A mutant version of the same plasmid (Fig. 4A) was virtually inactive, indicating that S897 integrity is essential to mediate EPHA2 effects (Fig. 4A). Similarly, the number of PC cells, stably transfected with BRAFV600E , was reduced by EPHA2 silencing and the cell number was partially rescued by wild-type EPHA2 but not its S897A mutant (Supplementary Fig. S6). Levels of total and phosphorylated EPHA2 upon knockdown and rescue transfections are shown in Figure 4B.
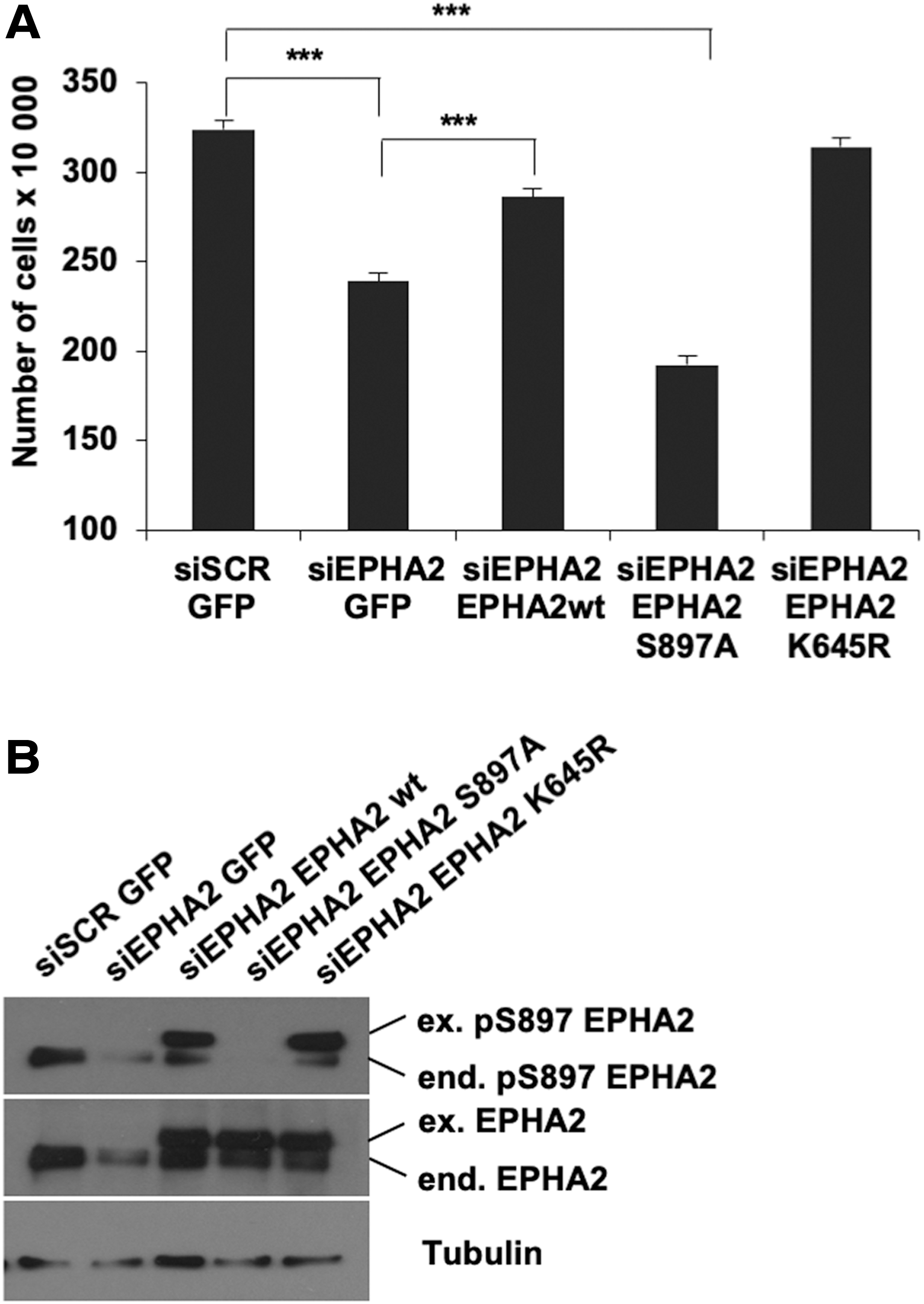
Effects of EPHA2 silencing on growth of BRAF mutant 8505C cells. EPHA2 expression was knocked down by siRNA transfection in 8505C cells. Control siRNA (siSCR) was used as negative control. Upon 36 hours of silencing, rescue of EPHA2 expression was obtained upon transfection with wild type (18), S897A mutant (S897A), or K645R mutant (K645R) EPHA2 vectors. GFP vector was used as a control; 36 hours after rescue, cells were counted (
It has been reported that S897 phosphorylation of EPHA2 regulates cell migration and invasion (28 –30). We therefore tested whether the migration of thyroid cancer cells was dependent on levels of pS897 EPHA2. Knockdown of EPHA2 reduced the ability of 8505C cells to close an artificial wound; this was rescued by transfection with wild-type EPHA2, but not with its S897A mutant (Fig. 5). To extend the analysis to more tumorigenic properties of cancer cells and cells harboring also the RET-PTC oncogene, we transfected TPC1, as well as 8505C cells, with siRNA targeting EPHA2 and tested migration (through wound healing assay) and Matrigel invasion (Supplementary Fig. S7A–D). We further performed cell count in TPC1, PC-PTC1, and PC-BRAF cells (Supplementary Fig. S7G–L), demonstrating significant impairment of growth and spreading of cancer cells upon silencing.
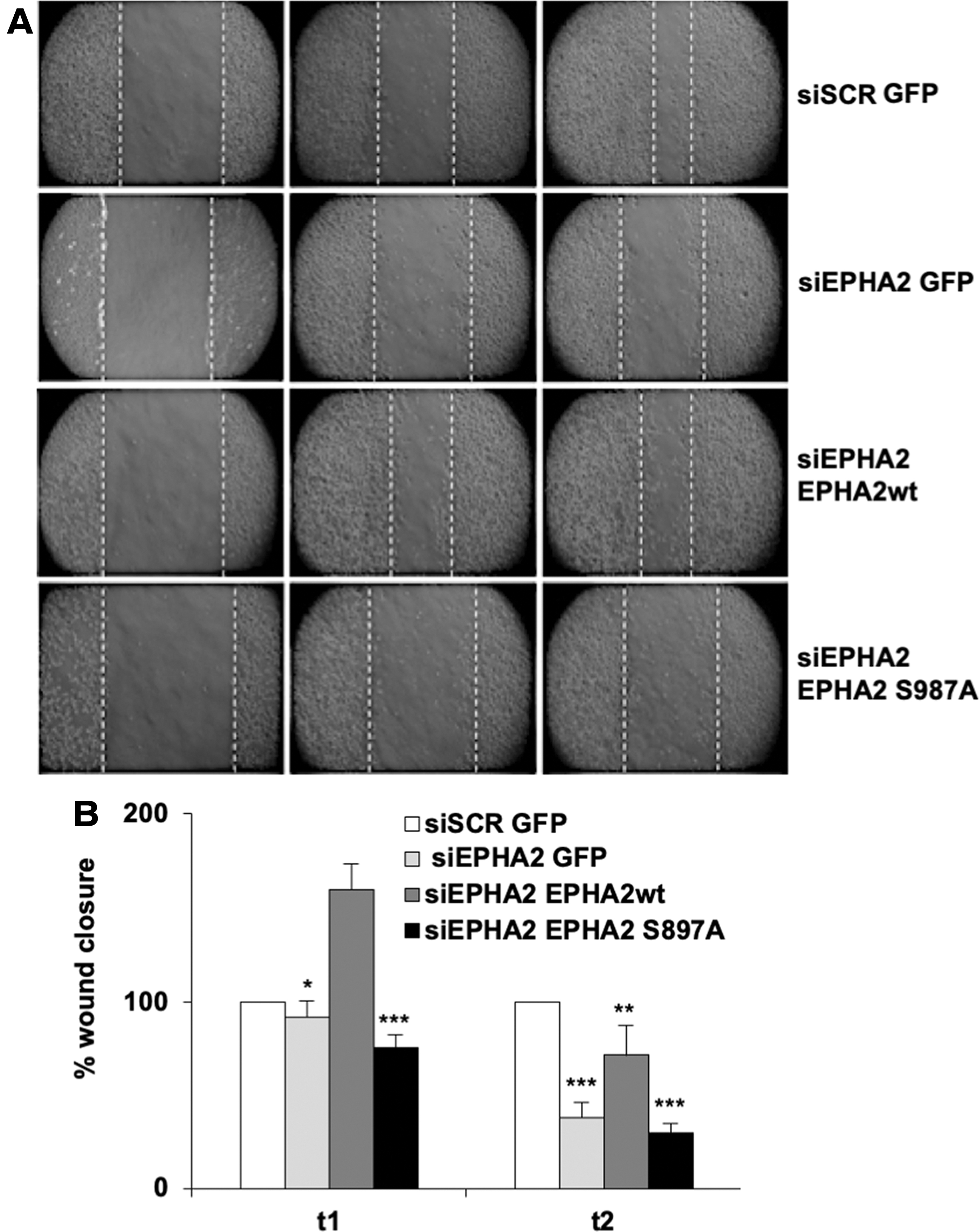
Effects of EPHA2 silencing on migration of 8505C cells. (
To then test the effects of EPHA2 stable silencing, we performed stable transfection of 8505C cells with shRNA targeting EPHA2 compared with scrambled control (shSCR). As shown in Figure 6, the silencing of EPHA2 in anaplastic thyroid cancer cells significantly affected cell growth (Fig. 6A) and the ability of cells to form colonies (Fig. 6B). Moreover, the reduced expression of EPHA2 had an important effect on the cells' ability to invade Matrigel (Fig. 6C, D) and to migrate (Fig. 6E, F), as well as to grow and spread in 3D Matrigel (Fig. 6G).
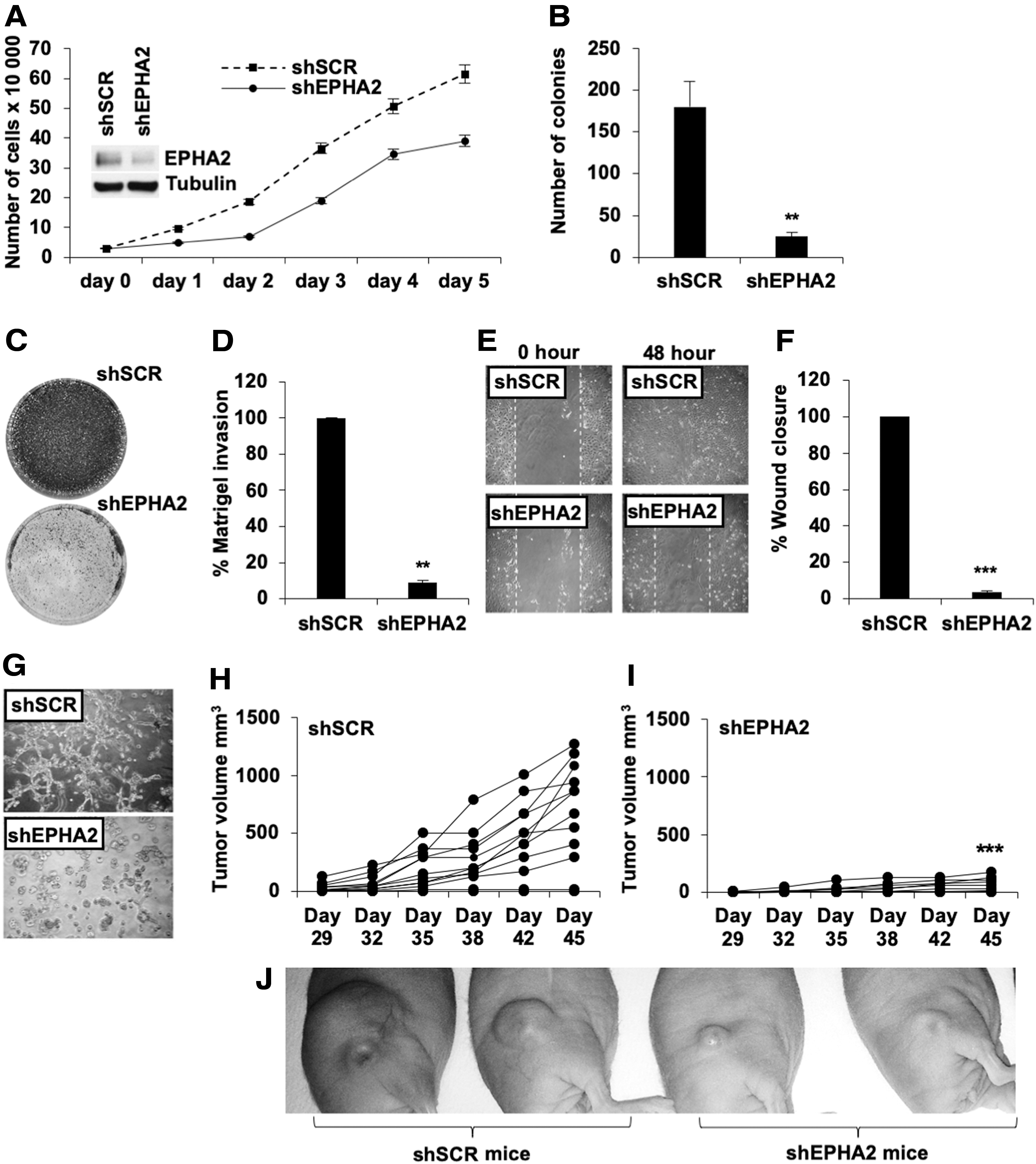
Effects of EPHA2 stable silencing on 8505C cells. (
To finally explore the in vivo tumorigenic properties of silenced cells, we performed xenograft transplantation of 10 × 106 8505C cells stably transfected with control (shSCR) or EPHA2 shRNA (shEPHA2) (Fig. 6H, I) and observed significant reduction in tumor growth in shEPHA2-transfected cells compared with the shSCR control (representative images of xenografts in shSCR and shEPHA2 mice are also shown in Fig. 6J). In conclusion, our results showed a significantly reduced tumor growth in shEPHA2-transfected animals, confirming an important role for EPHA2 in regulating the oncogenic properties of thyroid cancer cells.
Discussion
Here we show that EPHA2 phosphorylation on S897 is a novel hub in the ERK1/2 signaling network sustaining thyroid cancer cell proliferation and motility. Accordingly, the recent TCGA analysis of PTC has shown that ERK1/2 and RSK are robustly activated in PTC (5).
EPHA2 S897 is embedded in the consensus sequence for phosphorylation mediated by AGC serine/threonine kinases, that include AKT and p90RSK (35,39,40). RSK family kinases are prototypic effectors of the ERK1/2 signaling cascade mediating phosphorylation of the RSK C-terminal kinase domain (CTKD), consequently initiating a sequence of events leading to the activation of the RSK effector N-terminal kinase domain (NTKD) (35,40,41). By using chemical pathway inhibitors, we show that in BRAFV600E cells, EPHA2 S897 phosphorylation is selectively mediated by p90RSK. Instead, in RET-transformed cells not only p90RSK but also PI3K inhibition blunted S897 phosphorylation; this suggests that AKT is involved in this process, although it should be considered that through PDK1, PI3K can also contribute to full activation of p90RSK (41). Our in vitro kinase assays show that p90RSK1 is a bona fide S867 kinase and it requires both serine 897 and arginine 894 to efficiently phosphorylate S897-containing peptides.
We show that EPHA2 is robustly expressed and S897 is phosphorylated in both human thyroid cancer cells endogenously expressing RET fusions or mutant BRAF and RAS oncoproteins, and in rat thyroid cells adoptively expressing RET fusion or BRAFV600E (Supplementary Fig. S8). Consistent with findings by Miao (29) and Zhou (30), RNAi-mediated silencing combined with EPHA2 rescue experiments demonstrated that EPHA2 and its phosphorylation on S897 are involved in thyroid cancer cell proliferation and motility.
Signaling through EPH receptors can be ligand-independent or ephrin-dependent (13). Previous studies have shown that S897 phosphorylation is part of the ligand-independent EPHA2 mode of action (29). The exact mechanism through which S897 phosphorylation functions in intracellular signaling is still unclear. One possibility is that phospho-S897 acts by recruiting intracellular signaling transducers to EPHA2. Accordingly, Ephexin 4 interaction with EPHA2 is regulated by S897 phosphorylation. Ephexin 4 is a member of the Dbl-type guanine nucleotide exchange factor (GEF) family. It is able to promote cell proliferation and survival by activating RhoG and Rac small GTPases through the ELMO-Dock180 or ELMO-Dock4 complexes as well as the PI3K/AKT signaling pathway (27,42). On the contrary, it has been shown that S897 phosphorylation is favored by monomeric EPHA2 status.
In this context, while ligand-mediated EPHA2 oligomerization leads to increased tyrosine phosphorylation, cell contraction, and reduced tumor growth and motility, EPHA2 mutants that are unable to dimerize have increased motility promoting effects, S897 phosphorylation, and reduced tyrosine phosphorylation (43).
It is still unclear whether in ERK1/2-driven thyroid cancer cells, EPHA2 S897 phosphorylation impacts Rho/Rac and PI3K/AKT signaling, EPHA2 dimerization/tyrosine phosphorylation, or both. Elucidating this pathway will be crucial to devise strategies to intercept it to blunt thyroid tumorigenesis.
Footnotes
Acknowledgments
We gratefully acknowledge Dr. J.A. Fagin for the inducible PC cells, Dr. S.J. Gutkind for molecular constructs, and Dr. V. De Falco and F. Carlomagno for help with the kinase assay.
Author Disclosure Statement
The authors Allocca, Cirafici, Laukkanen, and Castellone declare no competing financial interests.
Funding Information
This study was supported by the Epigen-CNR flagship project, POR Campania FESR 2014–2020 “SATIN” grant, and POR Campania CUP B63D18000210007.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8