
Abstract
Background:
In humans, resistance to thyroid hormone (RTH) caused by mutations in the thyroid hormone receptor alpha (THRA) gene, RTHα, manifests as tissue-specific hypothyroidism and circulating thyroid hormone levels exhibit hypothyroid-like clinical features. Before the identification of patients with RTHα, several Thrα1 knock-in mouse models were generated to clarify the function of TRα1. However, the phenotypes of these mice were not consistent with the clinical presentation of RTHα in humans. For the present study, we generated an RTHα mouse model that carries the Thra1 E403X mutation found in human RTHα patients. Here, we report the gross phenotypes of this mouse RTHα model.
Methods:
Traditional homologous recombination gene targeting techniques were used to introduce a mutation (Thra1 E403X) in the mouse Thra gene. The phenotypes of the resulting mice were studied and compared with clinical features observed for RTHα with THRAE403X .
Results:
Thrα1E403X/E403X homozygous mice exhibited severe neurological phenotypes, such as spasticity and motor ataxia, which were similar to those observed in endemic cretinism. Thrα1E403X/+ heterozygous mice reproduced most clinical manifestations of patient with RTHα, such as a normal survival rate and male fertility, as well as delayed postnatal growth and development, neurological and motor coordination deficits, and anemia. The mice had typical thyroid function with a modest increase in serum triiodothyronine (T3) levels, a low thyroxine (T4)/T3 ratio, and low reverse T3 (rT3) levels.
Conclusions:
The Thrα1E403X/+ mice faithfully recapitulate the clinical features of human RTHα and thus can provide a useful tool to dissect the role of TRα1 in development and to determine the pathological mechanisms of RTHα.
Introduction
Thyroid hormones (TH) have important roles in human development and functional maintenance in adults (1,2). Biological activities of TH are mainly mediated through its binding to thyroid hormone receptors (THRs). As ligand-dependent transcription factors, THRs regulate expression of target genes by binding to TH response elements in the promoters of triiodothyronine (T3)-responsive genes. THRs are encoded by the thyroid hormone receptor alpha (THRA) and thyroid hormone receptor beta (THRB) genes, which each undergo alternative splicing to generate receptor subtypes (TRα1, TRα2, TRβ1, TRβ2, and TRβ3).
TRα1 is the major isoform in the brain, heart, bone, intestine, and skeletal muscle, whereas TRβ1 mainly functions in the liver, kidney, pituitary, and thyroid. Tissue resistance to the action of TH can arise from mutations in THRB or THRA that affect binding to TH and release of corepressors and recruitment of transcription coactivators that together result in dysregulation of genes targeted by TH (3,4).
Patients with mutations in THRA or THRB that produce resistance to thyroid hormone (RTH) syndromes termed RTHα and RTHβ, respectively, have different clinical features (5). RTHβ has been extensively studied, and the disease mechanisms are clearly described (6). RTHβ is characterized by elevated TH levels, with thyrotropin (TSH) concentrations that are either within the normal range or slightly elevated (4,5).
The first RTHα patient was identified in 2012 and was found to carry a THRAE403X mutation (7). The mutation results in a truncated TRα protein that lacks the C-terminal α-helix, H12 (7). In the absence of H12, TRα cannot stabilize TH in the ligand binding pocket, and the ability to recruit coactivator and release corepressor is lost. These features define THRAE403X as a dominant-negative mutation. The general clinical features of RTHα exhibit some characteristics of congenital hypothyroidism, such as growth and development retardation, motor and cognitive deficits, constipation, and macrocephaly. However, the hypothalamus–pituitary–thyroid axis is only minimally affected such that these patients have near-normal TH and TSH levels (8).
Most RTHα patients are heterozygous and have a mutation in the ligand-binding domain of TRα1. Among these mutations, several (A263S/V, L274P, D211G, N359Y) affect both TRα1 and TRα2 (9 –13). Similar to mutant TRβ1 receptors, mutant TRα1 receptors also show defective hormone-dependent activation and inhibit wild-type (Wt) TRα1 function in a dominant-negative manner. The clinical phenotypes can vary depending on the type of mutation. Patients with RTHα having frameshift and nonsense mutations present more serious phenotypes than those having missense mutations, which are believed to be associated with the ability of the mutant receptor to interact with transcription corepressors in the presence of TH (14,15). For clinical symptoms, age is a relevant factor for severity even among patients carrying the same mutation. For example, adults with some mutations (E403X, F397fs406X, D211G, and A263S) have milder phenotypes than children having the same mutation (7,11,12,16). The mechanism of this age-related improvement is currently unclear.
Before identification of the human THRA mutation, TRα1 knock-in mouse models (Thrα1R384C, Thrα1PV, Thrα1L400R, and Thrα1P398H) were generated to clarify the functions of TRα1, and these mice have provided insights into the important physiological roles of TRα1 (17 –20). However, the alternate splicing of the gene in the target vector was eliminated during design of the previous TRα1 knock-in mouse models (Thrα1R384C, Thrα1PV, and Thrα1L400R). This loss of splicing would result in overexpression of the mutant allele and exacerbate the phenotypes of heterozygous TRα1-mutant mice (18,20 –22). Therefore, compared with RTHα patients, the phenotype of these mice was more severe, particularly in terms of higher mortality and lower fertility relative to that seen in humans (17,20).
To clarify the role of TRα1 in development and adulthood, for the present study, we generated a new knock-in mouse model (Thrα1E403X) that corresponds to the nonsense mutation found in RTHα patient. The functional characterization of the THRA-E403X protein was described previously (7) and is similar to the previously reported mutations TRα1-PV and TRα1-L400R (17,20).
Interestingly, the heterozygous mutant mice (Thrα1E403X/+mice) exhibited most of the phenotypic features of THRAE403X/+ patient, including normal survival and fertility rate; delayed postnatal growth and development; neurological and motor coordination deficits; anemia; and typical thyroid function with a modest increase in serum T3, low total thyroxine (T4)/total T3 ratio, and low reverse T3 (rT3) levels. These results indicate that the Thrα1E403X/+ mouse is a faithful animal model of patients with RTHα.
Materials and Methods
Generation of the Thrα1E403X mouse model using a knock-in strategy
To generate the Thrα1E403X mouse model, traditional homologous gene targeting techniques was used. A bacterial artificial chromosome clone, RP23–395E10, harboring the mouse Thra gene, was ordered from Invitrogen (Carlsbad, CA). The targeting vector was constructed using a recombination system in Escherichia coli, as described elsewhere (23). Briefly, a vector containing a PGK/EM&-Neo cassette flanked by LoxP at both ends (5′ and 3′) was used to construct the targeting vector. A 12.6-kb Thra genomic DNA fragment containing exon 9 was inserted downstream of LoxP (Fig. 1). The mutation is located at nucleotide position 1725 corresponding to NCBI accession # NM_178060. The GAG codon coding for E was changed to the stop codon TAG to generate the THRA-E403X mutation. The mouse model was generated on a C57BL/6N background and bred with the same strain to produce homozygous and heterozygous animals that were identified by direct DNA sequencing.
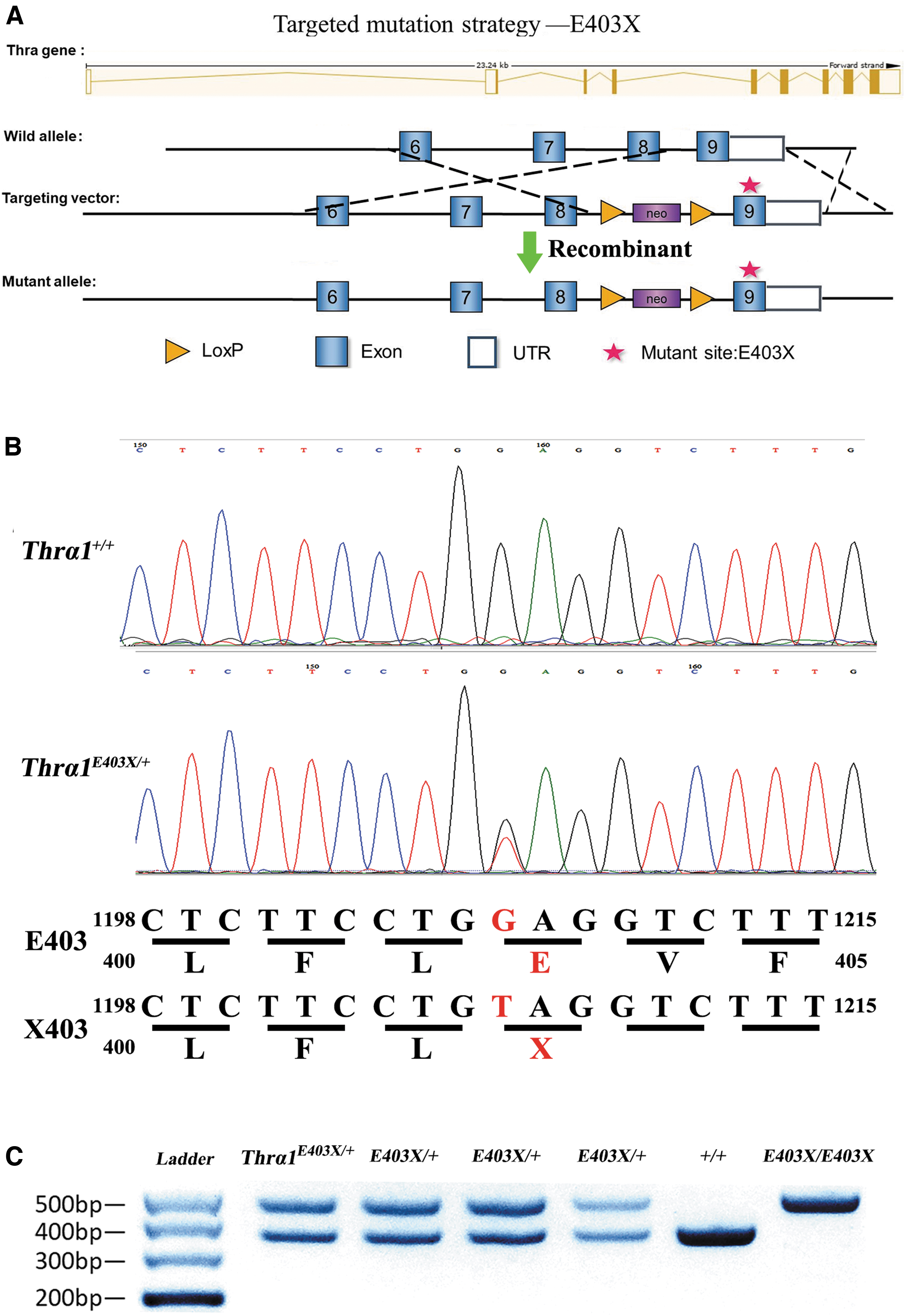
Generation of Thra1E403X
allele by homologous recombination. (
Animal maintenance
The studies involving animals were approved by the Animal Care and Use Committee at China Medical University and complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The mice were housed at 22°C ± 2°C with automatic 12-hour light and 12-hour dark cycles and free access to food and water. The Wt mice were weaned at 3 weeks of age, whereas the heterozygous and homozygous mice were weaned at 4–5 weeks due to delayed tooth rupture. Given the age-related difference in clinical features observed for RTHα patients, we analyzed the phenotypes of this mouse model at different ages that approximately correspond to ages in humans with 3-, 6-, and 16-week-old mice corresponding to human infancy, human puberty, and human adulthood, respectively.
Phenotype analyses
Bone analysis by micro-computed tomography
Femurs were analyzed using a micro-computed tomography (micro-CT) system (SkyScan 1276) operating at 50 kV and 200 μA, with a detection pixel size of 7 μm. Images were reconstructed using SkyScan NRecon software (Kontich, Belgium). In all groups, the left femur was excised and fixed in 10% phosphate-buffered formalin. Before scanning, the bones were transferred to 75% ethanol. The left femur was scanned to evaluate the overall structure and microstructure of the longitudinal and/or transverse sections. To build a three-dimensional (3D) bone model, a 0.215-mm region in the growth plate of the distal metaphysis was selected from a 1.72-mm region to determine the trabecular bone volume as a proportion of the tissue volume (bone volume [BV]/tissue volume [TV], an indicator of bone mass), trabecular number (Tb. N), and trabecular thickness (Tb. Th). In addition, based on a 0.43-mm-long region, the cortical thickness (Ct) and cortical BV/TV were determined from a 2.15-mm growth plate. The data set was used to construct the 3D bone image.
Grip strength
Muscle strength was determined using an automated mouse grip strength meter (Bioseb G3, Chaville, France). Mice were placed on a platform, allowed to grab a grid with either two forelimbs or all four limbs and then steadily pulled away until the mouse released the grid. Three trials were carried out to measure the forelimb strength, followed by three trials to measure the combined forelimb/hind limb grip strength.
Rota-rod treadmills
Mice were trained on Rota-rod treadmills (ENV-574M; Med Associates, Inc.). Before testing, mice were trained once daily on an accelerating rod for four consecutive days. On the fifth day, mice were allowed to run three times. Data were recorded for each run. The time on the Rota-rod was measured from the beginning of the trial (i.e., start of acceleration of rod rotation) until the mouse fell off the rod or onto the lever that stopped the timer. Mice were allowed to rest for at least 30 minutes between each trial.
Activity wheel
Mice were trained on an Activity Wheel (ENV-3046; Med Associates, Inc.) in a modular test chamber for the first day to allow adaptation to the environment. Testing was performed between the second and fifth day when the mice were allowed to run for three minutes daily. The data are presented as the average number of revolutions per mouse.
Body temperature
Rectal temperature was measured at 9:30 am each day for five days using a microprobe thermometer (BAT-12; Physitemp, Clifton, NJ) inserted 2–2.5 cm into the mouse rectum. The results are shown as the average value over five consecutive days.
Arterial blood pressure and heart rate
Resting mean arterial blood pressure and heart rate were measured using a noninvasive blood pressure monitor (BP-2010A; Softron, Beijing, China).
Assays for serum TH and insulin-like growth factor 1
Blood samples were collected from mice and centrifuged at 2580 g for 10 minutes at room temperature to collect serum samples, which were stored at −80°C before use. The serum levels of total T4 (TT4), total T3 (TT3), and rT3 were measured by radioimmunoassay (Beijing North Institute of Biotechnology, China). Serum TSH was determined using an enzyme-linked immunosorbent assay (ELISA) kit (SEA463Mu; Cloud-Clone Corp., Houston, TX). Serum insulin-like growth factor 1 (IGF-1) was measured using a mouse IGF-1 ELISA kit (EK0378; Boster, Wuhan, China).
Nissl and hematoxylin and eosin staining
Paraffin-embedded mouse tissues were sectioned into 5-μm-thick sections. Sections of the distal ileum and cerebral cortex were stained with hematoxylin and eosin and toluidine blue, respectively, using standard protocols.
Quantitative real-time polymerase chain reaction analysis of gene expression
Total RNA was extracted from the placenta using TRIzol Reagent (108-95-2; TaKaRa Bio, Inc., Kusatsu, Japan) following the manufacturer's instructions. cDNA was synthesized from 2 μg total RNA using a PrimeScript RT reagent kit (RR036A; TaKaRa). Quantitative real-time polymerase chain reaction was performed with a SYBR Premix Ex Taq Kit (RR820A; TaKaRa) and a LightCycler 480 Instrument (Roche Applied Science, Penzberg, Germany). The data were normalized to levels of GAPDH mRNA.
Statistical analyses
All statistical analyses were performed using SPSS software version 21.0 (IBM Corp., Armonk, NY). The results are expressed as mean ± standard error of the mean. The statistical significance of the differences was examined using two-tailed Student's t-test for nonpaired comparison. Comparison of the proportions was carried out with chi-square test. p-Values <0.05 were considered statistically significant.
Results
Function of the pituitary–thyroid axis
TT3 concentrations in Thrα1E403X/+ (Thrα1m/+) mice of either sex were slightly higher than those in Wt mice of the same age (Supplementary Fig. S1A). TT4 concentrations in male Thrα1m/+ mice were normal at 3 and 16 weeks, but lower at 6 weeks, compared with those in Wt mice. No significant differences in TT4 concentrations were observed between female Thrα1m/+ and Wt mice at each age (Supplementary Fig. S1B). A lower TT4/TT3 ratio and lower rT3 concentrations were observed for female Thrα1m/+mice compared with Wt mice in each age group. In contrast, in male Thrα1m/+ mice, lower TT4/TT3 ratios and lower rT3 levels were observed only for 3- and 6-week-old mice (Supplementary Fig. S1C, D). No significant differences in TSH levels were observed for either sex or any age between Thrα1m/+ and Wt mice (Supplementary Fig. S1E). As expected, TT4 and TT3 levels in Thrα1m/m mice were significantly lower than those in Thrα1m/+ and Wt mice of both sexes (data not shown).
Fertility and mortality
Thrα1m/+ mice were fertile. Male Thrα1m/+ mice had similar fertility to Wt mice. However, female Thrα1m/+ mice were less fertile than Wt female mice. Female Thrα1m/+ mice had an average of 5.71 pups across 24 pregnancies (Supplementary Table S1). The Thrα1m/m mice did not survive longer than 30 days with premature death occurring when the mice were between 21 and 30 days of age. The mortality rate of Thrα1m/+ mice was not significantly different from Wt mice.
To further investigate the reason for the low fertility of female Thrα1m/+ mice, we analyzed the placenta at embryonic day 13.5. The embryo absorption rate of pregnant Thrα1m/+ mice was significantly higher than that of Wt mice (17.78%, 8 of 45 embryos vs. 3.09%, 3 of 97 embryos, p = 0.012; Fig. 2-2A). Both the placental weight and fetal weight were lower in pregnant Thrα1m/+ mice compared with Wt mice (Fig. 2-2B, C). We analyzed genes associated with placental development including placental growth factor (PGF), vascular endothelial growth factor (VEGF), matrix metalloproteinase 2 (MMP2) and MMP9, GLUT1, GLUT4, Ki67, c-fos, c-jun, anti-inflammatory NOS2, and major apoptosis factor caspase3 and 9, as well as Dio2 and Dio3. The mRNA level of Ki67 was reduced by 43% (p < 0.05) compared with Wt mice, indicating impaired DNA duplication in Thrα1m/+ placental development (Fig. 2-3A). The expression of c-fos and c-jun, associated with differentiation and trophoblastic proliferation, was also reduced by 37% and 25% (p < 0.05), respectively, indicating that placenta growth was impaired (Fig. 2-3B, C). The amount of VEGF, which is associated with fetal vessels and dilation of maternal venous sinuses in the placental labyrinth, was decreased by 25% (p < 0.05) compared with Wt mice (Fig. 2-3D). Reduced amounts of VEGF are reported to be a main cause of abortion and fetal growth restriction (24). The mRNA levels of Dio2 and Dio3 were also significantly lower compared with Wt mice. This outcome is consistent with high levels of T3 and high T3/T4 ratios in Thrα1m/+ mice (Fig. 2-3E, F).
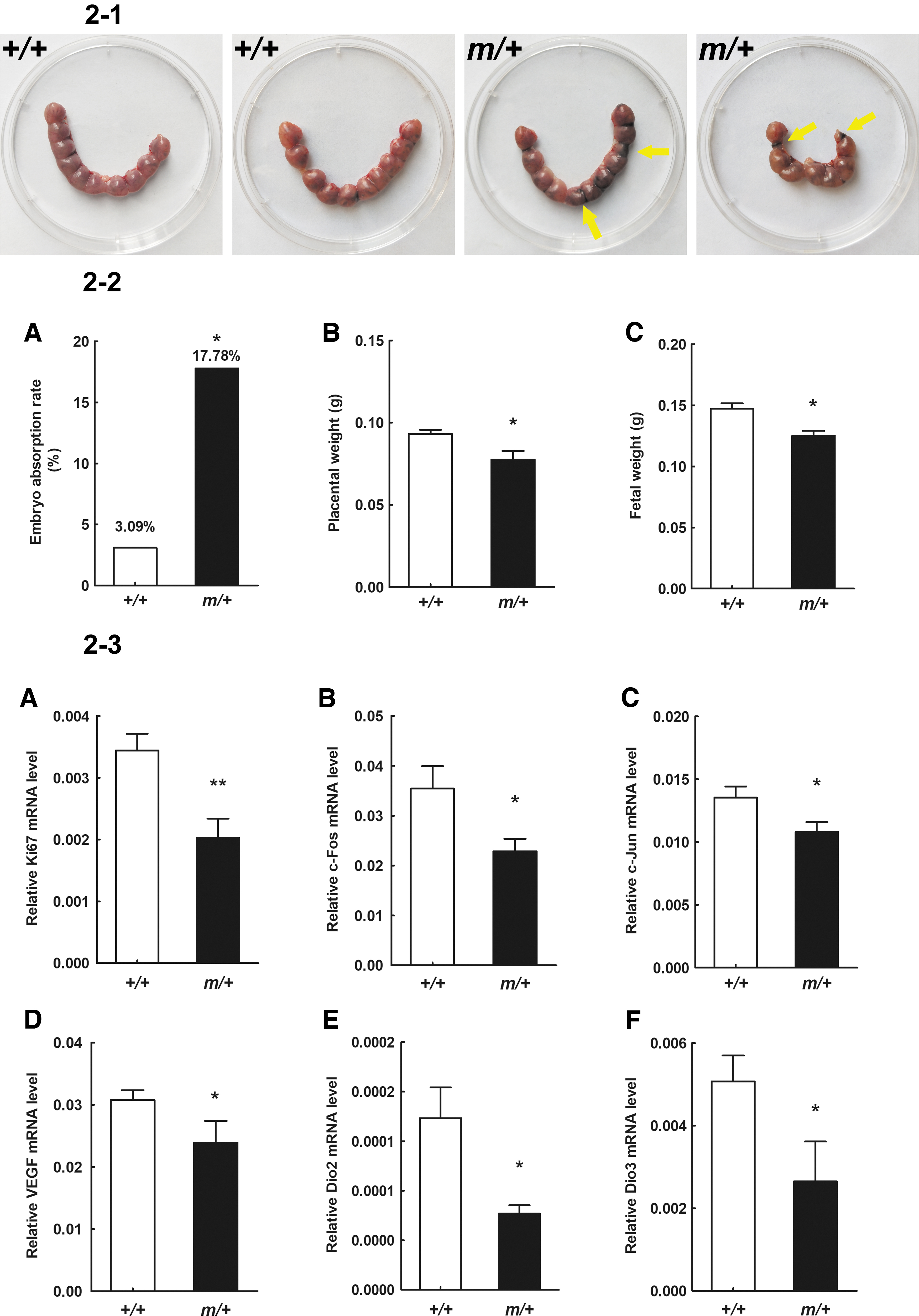
Placental and fetal development. (
Postnatal development and growth
The THRA-E403X mutation in mice was associated with retarded postnatal development and slowed growth of juveniles in both male and female mice. From puberty to adulthood, the Thrα1m/+ mice gradually made gains in body weight and length to reach levels close to those observed for the Wt mice (Fig. 3-1).

Postnatal development and growth of mice. (
Since the Thrα1m/m mice died before 30 days of age, we could only compare the growth of Thrα1m/m mice with Wt and heterozygous mice for 3-week-old mice. Male Thrα1m/m pups weighed 65.2% lower than Wt mice (3.82 ± 0.19 g [n = 6] vs. 10.98 ± 0.40 g [n = 8], p < 0.001) and 54.1% lower than Thrα1m/+ mice (3.82 ± 0.19 g [n = 6] vs. 8.33 ± 0.48 g [n = 10], p < 0.001; Fig. 3-2A). The body length of the Thrα1m/m male mice was 4.55 ± 0.14 cm (n = 6), which was 35.7% shorter than that of Wt mice (7.08 ± 0.11 cm [n = 8], p < 0.001; Fig. 3-2C) and 29.7% shorter than that of Thrα1m/+ mice (6.47 ± 0.08 cm [n = 10], p < 0.001; Fig. 3-2C). The tail length of the Thrα1m/m male mice (2.60 ± 0.17 cm [n = 6]) was 54.5% shorter than that of Wt mice (5.72 ± 0.22 cm [n = 8], p < 0.001) and 42.1% shorter than that of Thrα1m/+ mice (4.49 ± 0.17 cm [n = 10], p < 0.001; Fig. 3-2E).
Similar growth impairment was observed for female mutant mice. At 3 weeks old, the mean weight of Thrα1m/m female pups was 3.93 ± 0.19 g (n = 6), which was 60.7% lower than that of Wt mice (10.00 ± 0.62 g [n = 10], p < 0.001) and 49.7% lower than that of Thrα1m/+ mice (7.81 ± 0.63 g [n = 9], p = 0.018; Fig. 3-2B). The mean length of Thrα1m/m female mice was 33.1% and 25.2% shorter than that of Wt and Thrα1m/+ mice, respectively (4.60 ± 0.21 cm [n = 6] vs. 6.88 ± 0.15 cm [n = 10], p < 0.001, and 6.15 ± 0.15 cm [n = 9], p = 0.004; Fig. 3-2D). The tail length of Thrα1m/m female mice was 48.0% and 39.3% shorter than that of Wt and Thrα1m/+ mice, respectively (2.80 ± 0.21 cm [n = 6] vs. 5.38 ± 0.23 cm [n = 10], p = 0.000, and 4.61 ± 0.18 cm [n = 9], p = 0.02; Fig. 3-2F). Although the mechanism by which the mutation causes premature death of homozygous mice is unclear, the severe growth retardation associated with this mutation is likely one of the causes.
When Thrα1m/+ mice were 6–8 weeks old, significant reductions in body weight, body length, and tail length were observed. However, when Thrα1m/+ mice were 8 weeks old, they had gradually caught up in terms of growth and showed no significant difference above the age of 12 weeks compared with Wt mice (Fig. 3-1). These results suggested that delayed skeletal growth in Thrα1m/+ mice can result in body length differences at a young age. Interestingly, delayed skeletal growth is also observed in RTHα patients during childhood (7,16).
Skeletal phenotype
The Thrα1E403X mutation resulted in postnatal growth retardation in juvenile mice, although this growth retardation was largely recovered by the time Thrα1m/+ mice reached adulthood. To further explore bone development in Thrα1-mutant mice, we used 3D micro-CT to analyze the morphology and structure of long bones (femur) and flat bones (skull) of mice at different ages.
Micro-CT analysis showed that 3-week-old male Thrα1m/m mice had severe skeletal dysplasia. For example, no growth plate was formed and fewer bone trabeculae were present in the distal metaphysis (Fig. 4-1C, J), which is in line with the reduced bone mass seen for the femur (Fig. 4-2A). Compared with Wt mice, 3-week-old male Thrα1m/+ mice showed severe developmental delay of the epiphyseal distal femur (Fig. 4-1B, I), in addition to reduced femur bone volume (BV/TV) (Fig. 4-2A). The trabecular bone volume (BV/TV) and number (Tb. N) were decreased, and trabecular separation (Tb. Sp) was increased. Similarly, cortical bone thickness and cortical bone volume (BV/TV) were also decreased (Fig. 4-2B, C, E, F, G), indicating delayed endochondral ossification of the long bone during infancy.
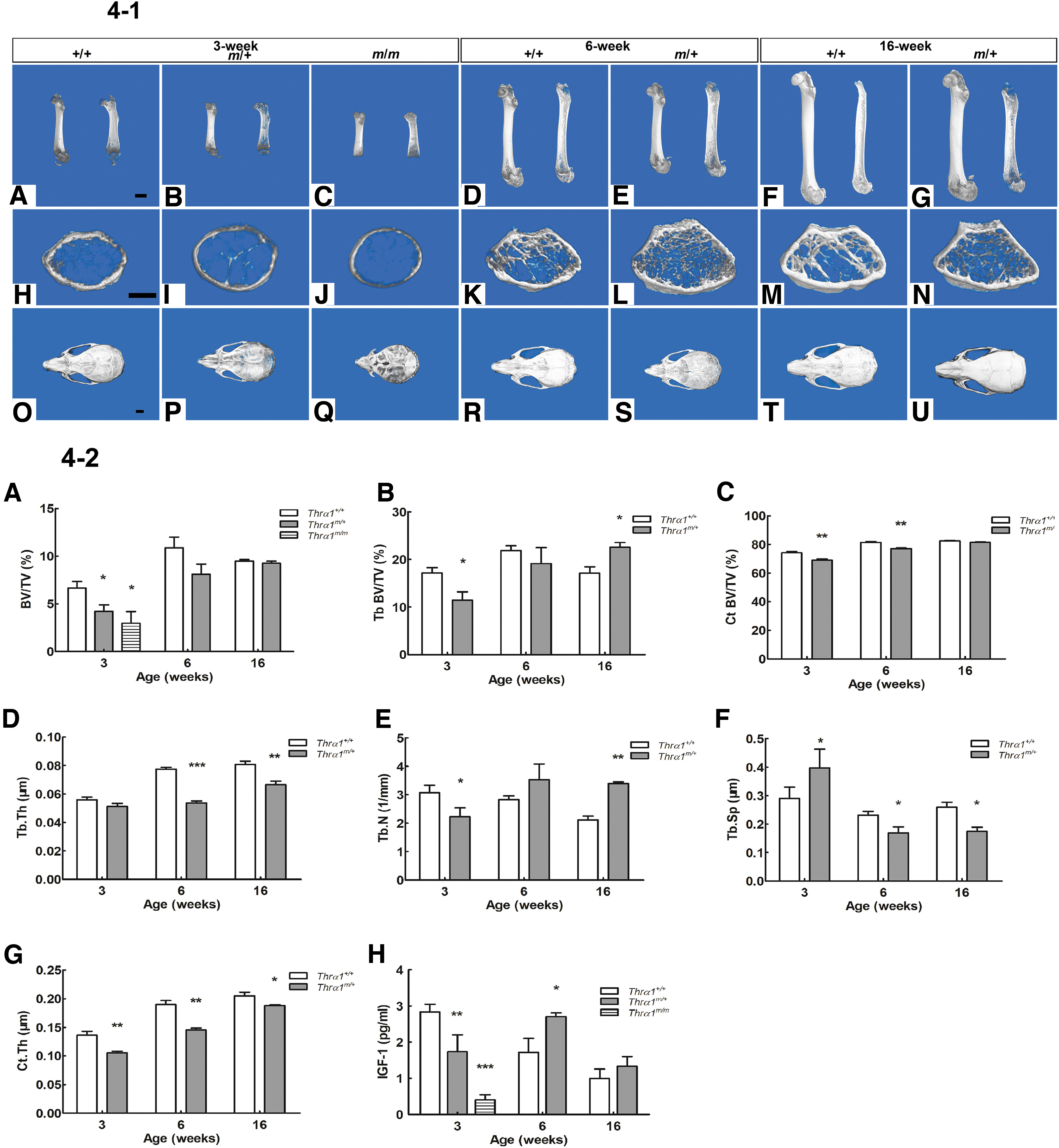
Bone development. (
Compared with 6-week-old male Wt mice, Thrα1m/+ mice had normal femoral bone volume (BV/TV). However, trabecular separation was decreased (Fig. 4-2A, F), indicating that “catch-up” growth of the femur occurred during puberty (Fig. 4-1D, E). Cortical bone thickness and cortical bone volume (BV/TV) were also decreased (Fig. 4-2C, G). By 16 weeks of age, Thrα1m/+ mice had normal femoral bone volume (BV/TV) as well as remarkable increases in trabecular bone volume (BV/TV) and bone number and decreased trabecular separation (Fig. 4-2A, B, E, F). These results are in line with the trabeculae accumulation and enlargement of the epiphyses in the distal femur (Fig. 4-1G, N), indicating abnormal osteoclast bone resorption associated with mutant TRα1. Despite the recovery in bone growth by 16 weeks of age, the femur length in Thrα1m/+ males was significantly shorter and the femur growth plates were wide compared with Wt mice, indicating a persistent delay in endochondral ossification (Fig. 4-1G).
Compared with 3-week-old Wt male mice, skull bones in both the Thrα1m/+ and Thrα1m/m mice were soft (Fig. 4-1P, Q), and the skull bones in Thrα1m/m mice were translucent and brittle, indicating a marked delay in intramembranous ossification of the skull. The fontanelles of 3- and 6-week-old Thrα1-mutant mice were larger, and the cranial sutures were wider than Wt mice, revealing a delay in fontanelle closure and suture fusion in juvenile mutant mice (Fig. 4-1P, Q, S). However, no difference in skull size was observed between the Wt and mutant mice (Fig. 4-1P, U).
IGF-1 is pivotal factor for normal longitudinal bone growth, skeletal maturation, and bone mass during development and bone maintenance in adult life. We measured the serum levels of IGF-1 in Wt and mutant male mice of different ages. At 3 weeks of age, serum IGF-1 levels were markedly reduced in Thrα1m/m male mice compared with Wt male mice. Interestingly, IGF-1 levels decreased for 3-week-old mice and increased in 6-week-old Thrα1m/+ male mice relative to Wt male mice of the same age, whereas the levels were similar when the mice were 16 weeks old (Fig. 4-2H).
Skeletal muscle strength
We assessed the muscle strength of the male mice using a grip test. The grip strength of all four limbs of 6-week-old Thrα1m/+ mice was 11% lower than that of Wt mice (155.11 ± 4.08 vs. 174.27 ± 5.17, p < 0.05). At 16 weeks of age, the difference between the four limb grip strength of Thrα1m/+ and Wt mice was essentially unchanged at 12%. Testing of forelimb grip strength showed a similar reduction (12%) for Thrα1m/+mice compared with Wt mice. These data suggest that muscle development of Thrα1m/+ mice was impaired and no age-dependent recovery occurred. In contrast, the body weight and body length of Thrα1m/+ mice was fully restored to the level of Wt mice when the mice were 16 weeks old. Bone development of Thrα1m/+ mice also partially recovered compared with Wt mice, but the recovery was not as complete as that observed for body growth.
Brain development
TRα1 accounts for 70–80% of the TRs in the brain. This receptor is expressed in all neurons and plays an important role in normal brain development (25,26). Information processing in the neocortex reportedly relies on a highly ordered cytoarchitecture and neuronal network to execute normal cognitive functions such as perception, voluntary movement, and language in humans (27). In the present study, we analyzed the cytoarchitecture of the cerebral cortex of TRα1-E403X mice. Figure 5-1 shows the cytoarchitecture of the cerebral cortex (A–I, × 100; J–R, × 40) on postnatal day (P) 0 (A–C; J–L), P10 (D–F; M–O), and P21 (G–I; P–R). Panels A–I show the cytoarchitecture of the primary somatosensory cortex that corresponds to the area enclosed by the rectangle in Panels J–R.
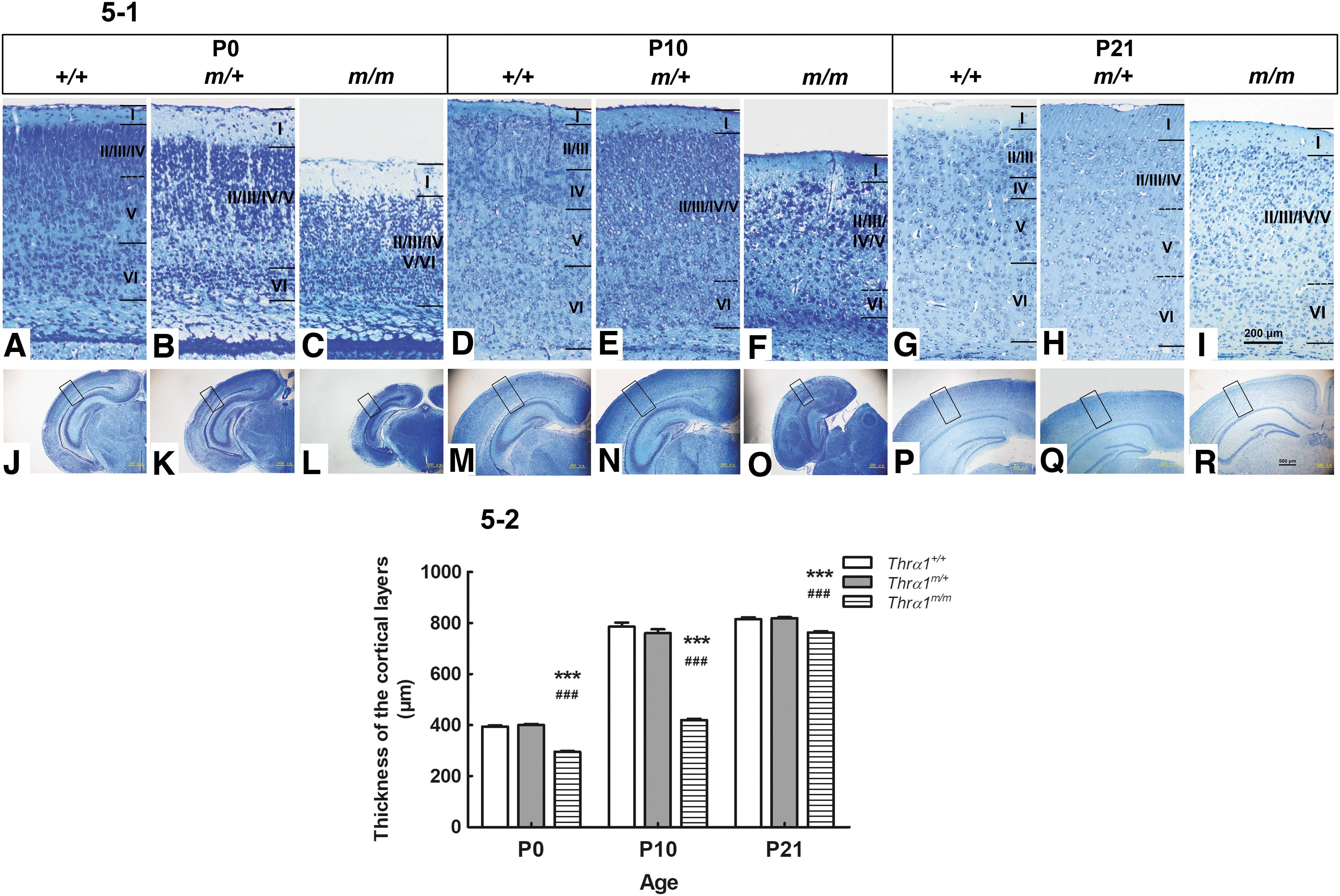
Representative photomicrographs of toluidine blue-stained coronal sections of the cerebral cortex. (
On postnatal day 0 (P0), both the inner granular layer (IV) and large pyramidal layer (V) had formed in Wt mice (Fig. 5-1A), whereas in Thrα1m/+ mice, layer V had not yet emerged (Fig. 5-1B). Thrα1m/m mice exhibited profound brain alterations, including a reduction in brain volume and thin cortical layers (Fig. 5-1C, 5-2), which persisted from P0 to P21.
Relative to Wt mice of the same age, for Thrα1m/+ mice at P10, the borders between layers II, III, IV, and V were indistinguishable, and large pyramidal neurons from layer V had entered layer IV and layer VI (Fig. 5-1E). In Thrα1m/m mice, the number of neurons was drastically reduced, a few patches of dense neurons were unevenly distributed, and the different types of neurons could not be distinguished in the cortical layers (Fig. 5-1F, O, and Fig. 5-2).
From P21 onward, the cortical cytoarchitecture of Wt mice was similar to that of adult mice, and the borders between the six cortical layers were clear (Fig. 5-1G, P). In Thrα1m/+mice, the borders between layers II, III, and IV were indistinguishable due to the entry of layer III pyramidal cells into layer IV in addition to the entry of layer V large pyramids into layer IV (Fig. 5-1H). Layers in Thrα1m/m mice showed reduced numbers of neural cells and a blurred border across the six layers (Fig. 5-1I, R, and Fig. 5-2) as well as unidentified types of neurons in each layer that indicated the influences of THRA-E403X-induced hypothyroidism on the cytoarchitecture of the cerebral cortex.
Thrα1E403X mice displayed a severe motor deficit (Supplementary Video SV1). Thrα1m/m males exhibited slow and clumsy movement in addition to impaired coordination and balance, which are similar to the motor ataxia observed in humans with congenital hypothyroidism. We also examined Thrα1m/+mice locomotor activity using Rota-rod treadmills and activity wheels. Compared with Wt mice, both male and female Thrα1m/+ mice had significantly reduced locomotor capacity at the age of 6 weeks. By 16 weeks of age, similar impaired locomotor activity was observed for males but not for females (Supplementary Fig. S2).
Internal organs
Compared with the Wt mice, both the heart weight and heart weight/body weight ratio of Thrα1m/+mice were reduced for juvenile (3 and 6 weeks) and adult (16 weeks) mice in both sexes (Supplementary Table S2). Although we could not measure arterial blood pressure and heart rate of 3- to 6-week-old mice given the small size of the heart at those ages, measurement of these parameters for 16-week-old mice showed no significant differences between the two groups (Supplementary Table S2).
The intestinal morphology showed obvious impairment in the length of the intestine (Supplementary Fig. S3-1A–C). In 3-week-old Thrα1m/m mice, the small and large intestine length was significantly shorter compared with Wt mice (p < 0.05) (Supplementary Fig. S3-2A–D). In Thrα1m/+ mice, only the large intestine length, but not that of the small intestine, was significantly shorter than Wt mice. By 6 weeks of age, Thrα1m/+ mice had normal intestine length. Interestingly, in older Thrα1m/+ mice (16 weeks old), the small intestine length was significantly longer for both males and females (9%, p < 0.01 and 5%, p < 0.05, respectively) compared with sex- and age-matched Wt mice. This phenomenon could be associated with changes in intestinal villi and crypts. To investigate this possibility, we performed histological analysis of the villi and crypts of the distal ileum. Intestinal development of 3-week-old Thrα1m/m males was severely impaired. The number of epithelial cells along the crypt–villus axis was reduced, and the length of the villi and the depth of the crypts were significantly reduced compared with age-matched Wt males (Supplementary Fig. S3-3C, 4A, B). In 3-week-old Thrα1m/+ male mice, the depth of the crypts was shorter, but the length of villi was similar to those in Wt mice (Supplementary Fig. S3-3B, 4A, 4B). At the age of 6 and 16 weeks, both the length of the villi and the depth of the crypts were decreased in Thrα1m/+ male mice (Supplementary Fig. S3-3E, G, 4A, B). Since villi length is associated with the absorptive function of the intestinal epithelium, and crypt depth is associated with the renewal ability of stem cells within the crypts and differentiation of epithelial cells that support the villi, we speculated that impairments in villi and crypts may affect nutrient absorption. Thus, the impaired growth and shorter life span of Thrα1m/m mice could be associated with this poor intestinal development and malnutrition.
The rectal temperature of Thrα1m/+ mice was nearly 1°C lower than that of the Wt mice at 6 and 16 weeks in both sexes (Supplementary Table S2). The inguinal white adipose tissue (iWAT), omentum majus white adipose tissue (omWAT), gonad white adipose tissue (gWAT), and intrascapular brown adipose tissue (iBAT) were measured when the mice were 3, 6, and 16 weeks old (Supplementary Fig. S4). At 3 weeks of age, the amount of the white adipose tissue (WAT), but not that of iBAT, in Thrα1m/+ male mice was significantly smaller compared with Wt mice. At 6 weeks of age, the amount of omWAT, but not that of other adipose tissues, was significantly reduced relative to Wt mice. At 16 weeks of age, there was no significant difference in all types of fat deposits between Wt and Thrα1m/+ male mice, indicating a delay in fat tissue development (Supplementary Fig. S4).
Blood system
Examination of blood from the mice, including red blood cell count (RBC), hemoglobin (Hb), and mean corpuscular volume (MCV), was performed for Wt and Thrα1m/+ mice at the age of 3, 6, and 16 weeks. RBC, Hb, and MCV levels in 3-week-old Thrα1m/+ mice (both male and female) were similar to those in Wt mice. The levels of RBC or Hb in both sexes of 6- and 16-week-old Thrα1m/+ mice were lower than those in Wt mice of the same age. The MCV was greater in Thrα1m/+males than in Wt mice at both 6 and 16 weeks. But for Thrα1m/+ females, the MCV was significantly greater only at 16 weeks (Supplementary Table S2). These data indicate that Thrα1m/+ males may have a higher frequency of macrocytic anemia at ages older than 6 weeks. For females, macrocytic anemia occurred at an older age (16 weeks) but not at young ages (6 weeks). Taken together, age-dependent anemia was observed for Thrα1m/+ mice. In humans, anemia is a clinical feature observed in some RTHα patients.
Discussion
The Thrα1E403X mouse is a novel TRα1 knock-in mouse model that mimics the clinical manifestation of RTHα mutation in humans. The main phenotypes of Thrα1E403X/+ mice are altered thyroid function (high T3, low or normal T4, low T4/T3 ratio, low rT3), anemia, delayed skeletal growth, and brain function abnormalities. These phenotypes recapitulate the clinical features of patients with RTHα, indicating that the Thrα1E403X/+ mouse model can serve as a useful tool to study human RTHα disease and treatment.
In contrast to the high mortality and low fertility rates observed for previous TRα1 knock-in mouse models of Thrα1PV/+ and Thrα1L400R/+, the Thrα1E403X/+ mice displayed a normal survival rate for both sexes, which makes the model easy to use for natural history studies. THs are important for the development and functions of the female reproductive system. Many studies demonstrated that the occurrence of hypothyroidism in women is associated with intrauterine growth retardation, spontaneous abortions, and infertility (28 –30). Our studies also demonstrated that maternal hypothyroidism caused a high rate of embryo absorption and abnormalities in fetal–placental development (31). Thrα1 is widely expressed in the syncytiotrophoblast and villous trophoblasts of the placenta (29,30). Our results indicated that the low fertility may be associated with abnormal placental development that leads to developmental defects in the embryos and subsequently termination of embryo development. Termination of the abnormal embryos could also halt growth of the placenta, although any one particular pregnancy may or may not have accompanying issues with embryo development. In the present study, we could not collect data for embryo development and thus could not examine how Thrα1 might affect embryogenesis. However, thra1 mRNA and protein are present at high levels in mouse embryonic stem (ES) cells where it can play a role in T3-mediated ES cell growth and differentiation (32,33). During embryogenesis of Xenopus, Thrα plays a critical role in controlling timing of developmental events and regulates the coordination of tissue-specific metamorphosis (34). In Xenopus, the effects of Thrα mutations on development vary depending on the location and type of mutation, and a similar variation is observed in human RTHα patients. Future in-depth study may shed more light on the role of RTHα in fertility.
In the present study, we found that skeletal bone growth in Thrα1E403X/+ juveniles exhibited characteristics similar to those seen for RTHα children, such as severe femoral epiphyses dysplasia and linear growth retardation. The femurs of adult Thrα1E403X/+ mice were short and thick, similar to those of adult patients with the THRAA382PfsX7 mutation, which is a similar mutation to THRAE403X, in that it involves a nonsense mutation that results in production of a truncated TRα1 protein. Our results confirmed earlier findings that TRα1 plays an important role in endochondral and intramembranous ossification and in balancing the processes of osteoclastic bone resorption and osteoblastic bone formation during postnatal growth (35,36).
Unlike the permanent dwarfism observed for Thrα1PV/+ and Thrα1L400R/+ mice, Thrα1E403X/+ mice showed delayed growth from the postnatal period 3 to 6 weeks of age. After 6 weeks of age, the mutant mice gradually caught up and reached a normal size by 16 weeks old, with the exception of a slight decrease in femur length. Our data suggested that TRα1 influences skeletal bone growth at early ages but not in mature adults. A similar skeletal phenotype was also reported for ThrαR384C/+ mice (18).
IGF-1 and GH are important factors that control bone growth and expression of genes that are mediated by TH (37,38). In the present study, compared with Wt mice, IGF-1 levels in Thrα1E403X/+ mice declined (3 weeks) and rose (6 weeks), before returning to normal Wt ranges (16 weeks). The transient increase occurs during a period that corresponds to the period when “catch-up” growth was observed. Our data indicated that IGF-1 is associated with growth retardation during infancy, which is supported by previous findings that TRα1 plays a dominant role in regulation of skeletal growth by mediating IGF-1 transcription from P0 to P3 weeks (39). Thrα1PV/+ mice also show a significant reduction in IGF-1, IGF-1 receptor, and GH receptor levels in bone growth plates (40). Since the “catch-up” growth we observed in the current study began when the mice were 6 weeks old, we hypothesize that TRα1 controls IGF-1 gene expression in infancy and other factors are involved in later skeletal bone development. Although IGF-1 levels and bone growth were increased at older ages, defects in bone formation such as osteoclastic resorption were not resolved.
Thrα1E403X/E403X homozygous mice exhibited severe neurological phenotypes, such as spasticity and motor ataxia, which are similar to those observed in endemic cretinism (41). Neuropsychological damage is one of the most prominent clinical features of patients with RTHα. Patients with RTHα carrying frameshift or nonsense mutations present with moderate neurological and motor coordinative impairments that are not improved by postnatal TH treatment (10,42,43). Meanwhile, RTHα patients with missense mutations presented with mild neuropsychological damage, which showed limited improvement with TH therapy (10,12,13). The phenotype for Thrα1E403X homozygous mice can serve as a platform for selection of potential treatment strategies for RTHα.
The intestine of TRα1E403X mice displayed significant elongation and impaired villi–crypt formation relative to Wt mice, and this phenotype is remarkably similar to that of TRα1PV mice (44). The elongated intestine may be due to impaired villi–crypt formation, and the abnormal villi could reduce nutrient absorption. Although extending the length of the small intestine may compensate for the functional reduction of the villi and allow nutrient absorption, this extended length will also increase the time needed for food passage and bowel emptying that could result in the constipation that is seen in RTHα patients. Further investigation of how intestinal dysfunction arises due to TRα1 mutation is needed.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by Chinese National Natural Science Foundation Grants 81970681 and 81570711 to X.T.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Video SV1