
Abstract
Approximately 5% of all cases of papillary thyroid cancer (PTC) are inherited. However, the susceptibility gene(s) for nonsyndromic familial PTC (FPTC) remain unclear. We performed whole genome sequencing of peripheral blood DNA samples from two affected family members with PTC. CHEK2 transcript expression and the protein levels of CHK2 and p53 were evaluated in the thyroid tissues from two affected members of the kindred and sporadic PTC cases. The entire CHEK2 coding sequence was examined by Sanger sequencing in blood DNA samples from 242 sporadic PTC patients. We identified a novel heterozygous germline mutation in CHEK2 (c.417C→A) that was detected in all available affected members of a kindred with FPTC. This variant was found in only 1 out of 264,200 persons in the Genome Aggregation Database and the NHLBI Trans-Omics for Precision Medicine program. The CHEK2 c.417C→A variant introduces a premature termination codon (Y139X). We found reduced CHK2 protein expression in tumor samples from the two patients who carried the variant as compared with sporadic cases without the variant. The Y139X loss-of-function variant led to reduced p53 phosphorylation and decreased p53 protein level. In addition, two rare missense variants (R180C and H371Y) in CHEK2 were identified in 5 (2%) of 242 patients with sporadic PTC. Our findings suggest that the CHEK2 Y139X variant may be associated with FPTC.
Introduction
The incidence of papillary thyroid cancer (PTC) has increased annually over the past 40 years (1). Approximately 5% of all cases of PTC are inherited (1 –3). Familial PTC (FPTC) is categorized into two groups: syndromic and nonsyndromic. Syndromic FPTC is associated with predominantly nonthyroid tumors, such as familial adenomatous polyposis, PTEN hamartoma (classic Cowden syndrome), Carney complex type 1, Werner's syndrome, and DICER1 syndrome, for which the susceptibility genes have been identified (3). The other group is nonsyndromic FPTC, in which the main feature is thyroid cancer; this form accounts for ∼95% of all the familial cases (3). Nonsyndromic FPTC is considered when two or more first-degree relatives are diagnosed with PTC, but the patient has no risk factors for thyroid cancer. The susceptibility genes for nonsyndromic FPTC are still unclear, although some low-penetrance susceptibility variants in NKX2-1, FOXE1, HABP2, and SRGAP1 and several candidate chromosomal loci have recently been described but most have not been validated in follow-up studies (4).
Materials and Methods
Study samples
This study was approved by the Research Ethics Board of the Beijing Hospital, Ministry of Health. All participants provided written informed consent before participation. Several members of the pedigree (Patients II.2, II.5, and II.6) were admitted to our hospital in 2018 due to thyroid neoplasms detected on ultrasonography and had ultrasound-guided fine-needle aspiration (FNA) biopsy (Table 1 and Fig. 1A). The proband (Patient II.2), a 38-year-old woman, was the oldest of four children. All three patients (Patients II.2, II.5, and II.6) were diagnosed with PTC and had thyroidectomy. One family member (Patient I.1) presented with a neck mass and hoarse voice and was diagnosed with PTC in 2013 at the age of 58 years. The age range of the family members in the third generation was 5 to 12 years old, and their blood samples were not available. None of the family members, except one member (Patient II.3), who died of nonsmall cell lung cancer 12 years ago, had a history of other primary cancers. No other benign tumors were detected in the family. Two hundred and forty-two unrelated Chinese individuals who were diagnosed with PTC based on histology after thyroidectomy were recruited into the study. None of the 242 patients had a family history of any type of cancer.

Identification of the Y139X variant of CHEK2 in a family with FPTC. (
Clinical Characteristics and Treatment in Family Members with Familial Papillary Thyroid Cancer
Staging was evaluated on the TNM classification of the AJCC and the Union for International Cancer Control (UICC), Eighth Edition.
Disease status was evaluated on the basis of follow-up ultrasonography and the serum thyroglobulin level.
Patient I.1 refused cytologic examination of a suspicious lymph nodes on ultrasonography.
Positive diagnosis of papillary thyroid cancer based on the pathologic evaluation.
AJCC, American Joint Committee on Cancer; NA, not applicable; TNM, tumor-node-metastasis; UICC, Union for International Cancer Control.
Whole-genome sequencing of blood samples and thyroid tissue samples
We performed whole-genome sequencing (WGS) of the peripheral blood DNA samples from Patients II.5 and II.6. The variants identified by WGS were validated in the affected and unaffected family members by Sanger sequencing. WGS was performed to profile somatic mutations in the tumors and matched cancer-free thyroid tissues from Patients II.5 and II.6. See details in the Supplementary Methods and Supplementary Tables S1 and S2.
Examining CHEK2 mutation in subjects with sporadic PTC
The germline genomic DNA of peripheral blood samples from 242 unrelated subjects with sporadic PTC was screened for CHEK2 mutation. The entire coding sequence (CDS) and splicing sites of CHEK2 were amplified by polymerase chain reaction (PCR), and then examined by Sanger sequencing. The primers used are listed in Supplementary Table S3.
Reverse transcription: PCR analysis
Total RNA was extracted from the thyroid tumor (T) and matched cancer-free thyroid tissues (N) from Patients II.5 and II.6 using Trizol (Life Technologies, Waltham, MA). Reverse transcription was performed using the M-MLV reverse system (Takara, Kusatsu, Shiga, Japan) to obtain complementary DNAs (cDNAs). An allele-specific PCR assay was performed using the cDNAs of the thyroid tumors (T) and matched thyroid tissues (N) from Patients II.5 and II.6 to examine the expression of mRNAs from wild-type allele and the mutant allele. The expression of EIF4H in thyroid tissue was used as a control (5). Primers used are listed in Supplementary Table S3.
Cell lines and cell culture
Two cell lines were used in the study. FTC-133 (Human follicular thyroid cancer cell line) was obtained from the Cell Resource Center, Peking Union Medical College, National Infrastructure of Cell Line Resource and human embryonic kidney (HEK) 293T cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). The identity of the cell lines was authenticated with short tandem repeat profiling. The cell lines were checked for mycoplasma contamination by PCR. FTC-133 cells were cultured in DMEM–Ham'F12 (1:1; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies),
Construction of plasmids and transient transfection
Wild-type CHEK2 transcript (NM_007194) was amplified from the cDNA of the affected family member (Patient II.5) by PCR. The Y139X mutation was introduced into the wild-type CHEK2 CDS clone using the overlapping PCR method. The CDS of wild-type and mutant CHEK2 was cloned into the pCDH-CMV-MCS-EF1 Lenti-vector (#CD513B-1; SBI, Palo Alto, CA), respectively. The HEK293T cells were seeded in a six-well plate and transfected with the wild type or mutant construct using Lipofectamine 2000 (Life Technologies) for 30 hours. The primers used are given in the Supplementary Table S3.
Protein blot analysis
Cell lines and tissue samples were lysed using radioimmunoprecipitation assay lysis buffer containing protease and phosphatase inhibitors (Roche, Basel, Switzerland), and were sonicated for 20 seconds. The lysate was resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA). Membranes were blocked for 1 hour and blotted for various primary antibodies overnight in 5% nonfat milk or 5% bovine serum albumin (BSA) in Tris buffered saline solution, 0.5% Tween-20 (TBST; Thermol Fisher Scientific, Waltham, MA). The following primary antibodies were used: anti-CHK2 (1:10,000, #ab109413; Abcam, Cambridge, United Kingdom, an antibody against a synthetic peptide [Human CHK2 aa 1-200]), anti-Phosphor-p53 (Ser 20, 1:1000, #sc-18079-R; Santa Cruz Technology, Dallas, TX), anti-p53 (1:1000, #sc-263; Santa Cruz Technology), and anti-beta-Actin (C4, 1:1000, #sc-47778; Santa Cruz Technology). Horseradish-peroxidase conjugated antibodies to mouse (1:5000, #ab6789; Abcam) or to rabbit (1:5000, #ab6721; Abcam) were used as the secondary antibodies, and Immobilon Western Chemiluminescence HRP Substrate (Millipore) was used for detection.
Immunohistochemistry
Thyroid tumor tissues and matched cancer-free thyroid tissues were fixed in 4% paraformaldehyde for 24 hours at room temperature. Consecutive paraffin sections of the corresponding tissues (3 μm thickness) were used for hematoxylin and eosin staining and immunohistochemical analyses. The primary antibody against CHK2 protein (#ab109413; Abcam, an antibody against a synthetic peptide [Human CHK2 aa 1-200]) was used at 1:400 dilution and incubated overnight at 4°C.
Results
Identification of the CHEK2 Y139X variant in FPTC.
We performed WGS of peripheral blood DNA from the affected family members with nonsyndromic FPTC (Patients II.5 and II.6) (Fig. 1A and Table 1). Using filtering criterion (Supplementary Fig. S1A and Supplementary Table S1), we identified two candidate protein-altering single-nucleotide variants that segregated with PTC in the family members. One variant (c.595G→A, p.G199R) was in the exon of TPO (Supplementary Fig. S1B). However, we did not further evaluate this variant because the affected family members had normal thyroid hormone levels when diagnosed with PTC (Supplementary Table S2). The other variant was in the CHEK2 gene, which encodes the checkpoint kinase CHK2 (6). The mutation results in a heterozygous single-base substitution in exon 3 of CHEK2 (chromosome 22: 29,121,258 C→A, rs200917541 [NM_007194:c.417C→A]). This CHEK2 c.417 C → A mutation was present in all affected family members with PTC (Patients I.1, II.2, II.5, and II.6) but was not in the unaffected spouse (I.2) (Fig. 1A, B and Table 1). In addition, this variant was absent in 138,632 exomes and whole genome sequences reported in the Genome Aggregation Database (7) and was only detected in 1 of 125,568 persons in the NHLBI Trans-Omics for Precision Medicine program. Furthermore, all affected family members were negative for previously implicated variants in FPTC (Table 1 and Supplementary Table S3). The C-to-A substitution converts a tyrosine residue to a premature stop codon (Y139X). Y139 is present within the forkhead-associated domain (FHA) of the CHK2 protein and is a highly conserved site not only in CHK2 but also in other FHA-containing proteins, such as SNIP1 and PPP1R8 (Fig. 1C), which are involved in carcinogenesis through transcriptional activation of c-MYC (8) and regulation of EZH2-mediated gene silencing (9), respectively.
Since the Y139X mutation creates a 5′ premature termination codon in the CHEK2 CDS, we examined the mRNA transcribed from the mutant allele in the thyroid tumor–normal pairs from Patients II.5 and II.6 using an allele-specific PCR and Sanger sequencing. The corresponding CHEK2 mRNA from the mutant allele was barely observed (Fig. 2A, B), and the protein product from the Y139X variant was not detected in both tumors and their matched tissues (Fig. 2C and Supplementary Fig. S1C), suggesting that the mutation triggers the nonsense-mediated mRNA decay (NMD), which results in a functionally null allele in the affected family members. We further assessed the CHK2 protein levels in the thyroid tumors from Patients II.5 and II.6 and subjects with sporadic PTC without the Y139X variant. We observed substantially lower CHK2 protein levels in the affected family members than those of sporadic cases (Fig. 3A, B and Supplementary Fig. S1D).

Expression of the Y139X variant of CHEK2 in thyroid tissues from the affected family members. (
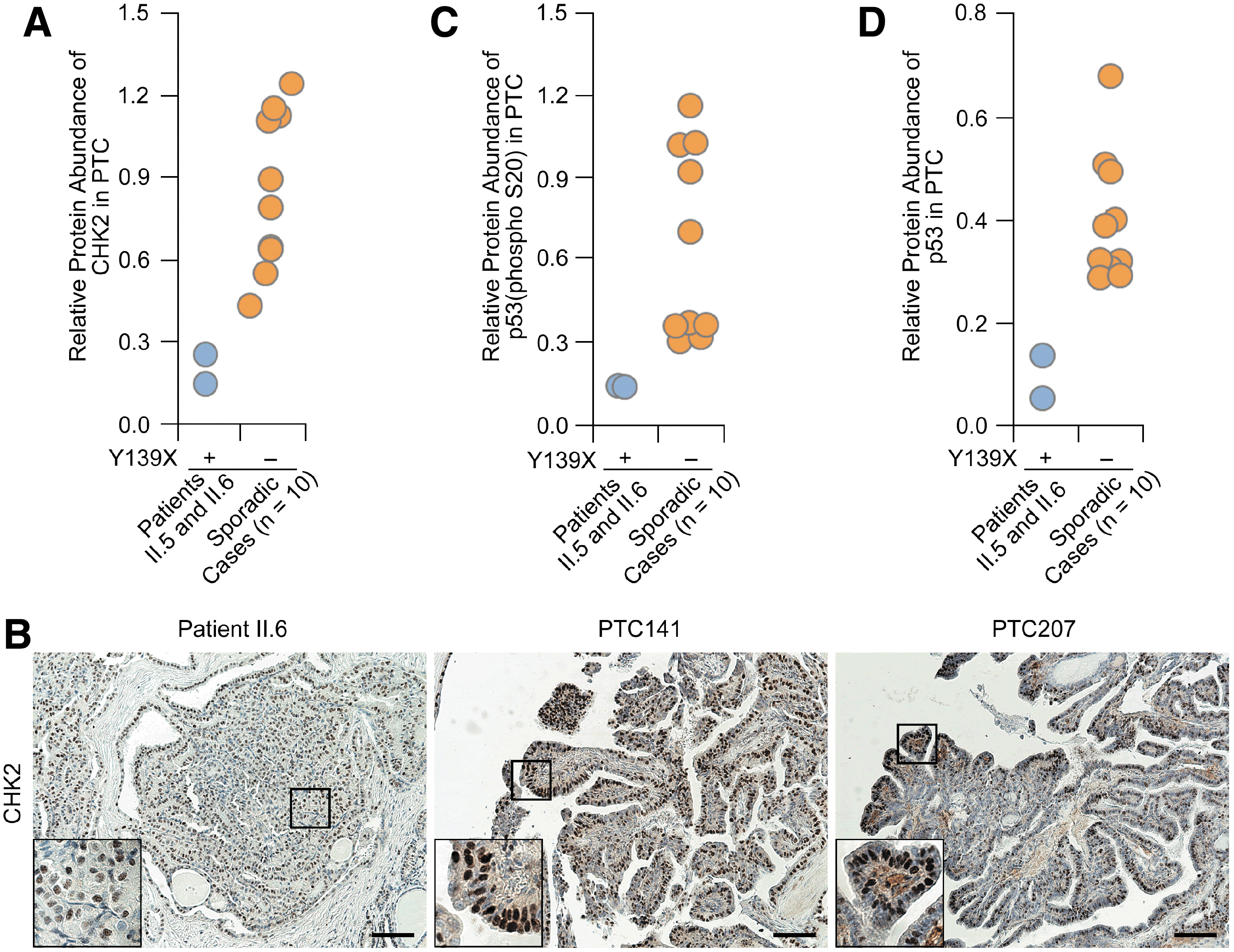
Decreased expression levels of CHK2, p53, and p-p53 in thyroid tumors from patients with FPTC. (
Functional characterization of the CHEK2 Y139X mutation
We next evaluated the functional consequence of reduced CHK2 expression due to the mutation. A critical role of CHK2 in response to DNA damage is to phosphorylate p53 on Serine20 (S20), which consequently promotes p53 protein stability through disruption of MDM2-mediated p53 degradation (10). We found decreased S20 phosphorylation levels and decreased p53 protein abundance in the thyroid tumors from Patients II.5 and II.6 as compared with those in sporadic PTCs without the variant (Fig. 3C, D and Supplementary Fig. S1D).
Among currently known mutations of thyroid cancer-related genes and pan-cancer drivers, only BRAFV600E was present in the two tumor samples, whereas none of these variations were observed in the matched thyroid tissues (Table 1 and Supplementary Fig. S2). Finally, we screened the entire CHEK2 CDS and splicing sites in the germline DNA from 242 sporadic PTC patients by Sanger sequencing. Only 2 rare missense variants (R180C in 2 cases and H371Y in 3 cases) were detected in these sporadic cases (Supplementary Fig. S3).
Discussion
To date, the link between CHEK2 and FPTC is unknown. Four low-penetrance CHEK2 variants (IVS2 + 1G>A, I157T, 1100delC, and del5395) have been reported to be associated with a moderately increased risk of sporadic PTC in the Polish population (11 –14).
Low-penetrance germline mutations of CHEK2 (R117G, R145W, delE161, and 1100delC) have been identified in human malignancies (15,16). Of them, the R117G, R145W, and delE161 mutant proteins had low abundance due to instability of the corresponding proteins (17). Mice with heterozygous1100delC truncating mutation, which abrogates the kinase of activity of Chk2, developed more spontaneous tumors than wild-type mice (18). In addition, Chk2-deficient mice were predisposed to UV-B irradiation-induced skin tumorigenesis, and Chk2-deficient cells showed deficiency in p53 phosphorylation on Serine20 and p53 stability (19,20). In this study, the Y139X mutation resulted in a strong decrease in mutant mRNA in the affected family members, suggesting that the NMD pathway may be triggered. NMD-triggered loss-of-function causes disease owing to haploinsufficiency, as exemplified by the generation of premature stop codons in the SOX10 gene, causing Waardenburg syndrome (21). Thus, our results and others' data already mentioned suggest that some CHEK2 mutants may contribute to tumorigenesis through the haploinsufficiency mechanism due to low CHK2 protein levels.
Our study highlights DNA repair deficiency as a potential mechanism driving FPTC susceptibility. In addition, whether the Y139X variant results in cases of FPTC in other families and confers a predisposition to other hereditary cancers needs to be investigated. Together, our study provides the first evidence identifying that CHEK2 Y139X variant may be associated with FPTC. Our findings will contribute to the understanding of FPTC.
Footnotes
Acknowledgments
We thank the family for participating in the project, Jiawei Wang from WuXiNextCODE for advice in analyzing WGS data, and Bayi Xiao for work with the samples collected for papillary thyroid cancer in Beijing Hospital.
Author Disclosure Statement
No competing financial interests exist.
Author Contributions
Drs. Miao and Zhu had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Zhu, Miao, and Zhao conceptualized and designed the study. All authors acquired, analyzed, or interpreted the data. Zhu, Zhao, and Miao drafted the article. All authors critically revised the article for important intellectual content. Zhu, Miao, Zhao, Yu, Cheng, and Wang provided administrative, technical, or material support.
Funding Information
This study was supported by grants from the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (2018-I2M-1-002) (to Dr. Zhao); Beijing Natural Science Foundation (7172194) (to Dr. Miao); and National Natural Science Foundation of China grants (81872096) (to Dr. Zhu), (81541152) (to Dr. Zhao).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3