
Abstract
Background:
Mutations of the thyroid hormone receptor α (THRA) gene cause resistance to thyroid hormone (RTHα). RTHα patients exhibit very mild abnormal thyroid function test results (serum triiodothyronine can be high-normal to high; thyroxine normal to low; thyrotropin is normal or mildly raised) but manifest hypothyroid symptoms with growth retardation, delayed bone development, and anemia. Much has been learned about the in vivo molecular actions in TRα1 mutants affecting abnormal growth, bone development, and anemia by using a mouse model of RTHα (Thra1PV/+ mice). However, it is not clear whether TRα1 mutants affect lymphopoiesis in RTHα patients. The present study addressed the question of whether TRα1 mutants could cause defective lymphopoiesis.
Methods:
We assessed lymphocyte abundance in the peripheral circulation and in the lymphoid organs of Thra1PV/+ mice. We evaluated the effect of thyroid hormone on B cell development in the bone and spleen of these mice. We identified key transcription factors that are directly regulated by TRα1 in the regulation of B cell development.
Results
: Compared with wild-type mice, a significant reduction in B cells, but not in T cells, was detected in the peripheral circulation, bone marrow, and spleen of Thra1PV/+ mice. The expression of key transcription regulators of B cell development, such as Ebf1, Tcf3, and Pax5, was significantly decreased in the bone marrow and spleen of Thra1PV/+ mice. We further elucidated that the Ebf1 gene, essential for lineage specification in the early B cell development, was directly regulated by TRα1. Thus, mutations of TRα1 could impair B cell development in the bone marrow via suppression of key regulators of B lymphopoiesis.
Conclusions:
Analysis of lymphopoiesis in a mouse model of RTHα showed that B cell lymphopoiesis was suppressed by TRα1 mutations. The suppressed development of B cells was, at least in part, via inhibition of the expression of key regulators, Ebf1, Tcf3, and Pax5, by TRα1 mutations. These findings suggest that the mutations of the THRA gene in patients could lead to B cell deficiency.
Introduction
The genomic signaling by thyroid hormones in growth, development, differentiation, and metabolic homeostasis is via thyroid hormone nuclear receptors (TRs). Two human TR genes, THRA and THRB, encode three thyroid hormone-binding receptor isoforms (α1, β1, and β2) (1). The expression of TR isoform is tissue-dependent and developmentally regulated (2). In the past decades, much has been learned about the TR actions in the major target tissues, such as the liver, brain, bone, and heart. The actions of TR in the hematopoietic organs have just begun to be explored. Interest in understanding the role of TRα1 in hematopoiesis was spurred by the findings that patients with mutations of the THRA gene manifested erythroid disorders (e.g., anemia) as one symptom of resistance to thyroid hormone (RTHα).
Using a mouse model of RTHα, the Thra1PV/+
mouse, Park et al. showed how a TRα1 mutant (TRα1PV) causes anemia (3). TRα1PV is a dominant negative mutant, sharing mutated sequences in the C-terminal truncation mutation (398-PPFVLGSV
The availability of the mouse model of RTHα, the Thra1PV/+ mouse, provided us with an opportunity to ascertain whether TRα1 mutants could lead to defective lymphopoiesis in patients. Previous studies had shown that mice deficient in TRα1 had a decreased proliferation of B cell progenitors and reduced normal B cell pool (6). The present studies aimed to elucidate how TRα1 mutants could affect primary B cell development in Thra1PV/+ mice. We found a significant reduction in B lymphocytes in the peripheral circulation, bone marrow, and spleen. The expressions of key regulators of B cell development, such as Ebf1, Tcf3, and Pax5, were significantly decreased in the bone marrow and spleen of Thra1PV/+ mice. We further elucidated that the Ebf1 gene, a transcription factor essential for lineage specification in early B cell development, was directly activated by triiodothyronine (T3) mediated by TRα1. Thus, mutations of TRα1 could impair B cell development in the bone marrow via suppression of key regulators of B lymphopoiesis.
Materials and Methods
Mice and treatment
All animal studies were performed according to the approved protocols of the National Cancer Institute Animal Care and Use Committee. The animal study protocol is NCI LMB-036. Generation of Thra1PV/+ mice was previously described (7). To induce hypothyroidism, 4- to 6-month-old female mice were fed a low-iodine diet supplemented with 0.15% propylthiouracil (LoI/PTU) (Cat# TD 95125; Harlan Teklad, Madison, WI) for 10 days. To induce hyperthyroidism, mice fed with PTU diet were injected with T3 (5 μg per mouse) intraperitoneally daily for six days (T3, Cat# T2752; Sigma–Aldrich, St. Louis, MO).
Peripheral blood profile analysis
Peripheral blood was collected in a heparinized microtube and analyzed by hematology analyzer (Hemavet HV950FS; Drew Scientific, Miami Lakes, FL).
Flow cytometry analysis
Peripheral blood mononuclear cells were collected by using Histopaque-1083 (Sigma–Aldrich) according to the manufacturer's instructions. Single-cell suspensions from the bone marrow and spleen were prepared as described previously (8). Flow cytometry analysis was carried out as previously described (3). Supplementary Table S1 lists the antibodies with clone IDs used in flow cytometry analyses. All antibodies were purchased from eBioscience for fluorescence-activated cell sorting analyses.
RNA extraction and quantitative reverse transcription–polymerase chain reaction
Total RNA was isolated from the bone marrow and spleen using TRIzol (Thermo Fisher Scientific, Waltham, MA). Quantitative reverse transcription–polymerase chain reaction (RT-qPCR) and quantitative analyses were performed as previously described (3).
The primer sequences are listed in Supplementary Table S2.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay of bone marrow cells was performed as described previously (9). Monoclonal antibody against TRα1, C4, used in the ChIP was developed in-house (10). To quantify the amount of precipitated DNA and further detect the promoter region of Ebf1, real-time qPCR was conducted using chromatin DNA. The primer sequences are shown in Supplementary Table S2.
Construction of luciferase reporters
The Ebf1 luciferase reporter cloned in pLG3 plasmid (Ebf1-Luc) was provided by Dr. Mikael Sigvardsson (Lund University, Sweden). For the construction of truncated Ebf1 Luc reporters, truncated DNA fragments of the Ebf1 promoter were prepared by using standard molecular cloning methodology upon request. The luciferase reporters were confirmed by restriction enzyme mapping and further validated by Sanger DNA sequencing analysis. The primer sequences are shown in Supplementary Table S3.
Luciferase reporter assays
Ebf1-Luc reporter plasmids, with or without the expression plasmid for TRα1, were transfected into CV1 cells using lipofectamine. Lysates prepared from transfected cells with or without T3 (100 nM) were assayed for luciferase activity, which was normalized to total protein concentration. Transfection experiments were repeated at least three times.
Western blot analysis
Western blot analysis was performed as previously described (9). Primary antibodies for TCF3 (sc-416), EBF1 (ab108369), PAX5 (8970S), and GAPDH (2118S) were purchased from Santa Cruz Biotechnology (Dallas, TX), Abcam (Cambridge, MA), and Cell Signaling Technology (Danvers, MA), respectively. Band intensities were analyzed by densitometry, and the density values were normalized to the GAPDH and quantified by using the National Institutes of Health imaging software (ImageJ 1.48v).
Statistical analyses
Student's t test was used for statistical analysis in the study. All statistical analyses were performed, and statistical significance was set at p < 0.05. All data are expressed as mean ± standard error of the mean.
Results
Decreased lymphocytes in the peripheral blood of Thra1PV/+ mice
Peripheral blood analysis shows that indices for white blood cells and lymphocytes were significantly decreased 10% and 17%, respectively, in Thra1PV/+ mice versus wild-type (WT) mice (Fig. 1Aa, b). We also found that total cell numbers in the bone marrow, spleen, and thymus were decreased 39%, 46%, and 25%, respectively, in Thra1PV/+ mice versus WT (Fig. 1B[a–c]). Using the markers and gating strategies reported by others (6,11,12), we found that B220+ B cells were decreased 59%, 43%, and 23% in the peripheral blood (Fig. 1C[I]), bone marrow (Fig. 1C[II]), and spleen (Fig. 1C[III]) of Thra1PV/+ mice, respectively (Fig. 1C[I–III]a). In contrast, no significant differences were detected in the number of T cells in the peripheral blood, bone marrow, spleen, and thymus between WT and Thra1PV/+ mice (Fig. 1C[I–III]b). The respective dot blots and the gating strategies are shown in Supplementary Figure S1. Using CD19+ and B220+CD19+ as markers for B cells, we demonstrated that the extent of reduction of B cell population in the blood, bone marrow, and spleen of Thra1PV/+ mice was similar to those using B220+ as marker alone (Supplementary Fig. S2). The number of macrophages and granulocytes that composed of white blood cells was not significantly changed in Thra1PV/+ mice versus WT mice (data not shown). These results indicated that mutation of the Thra1 gene specifically affected the lymphocyte development in the B cell lineage.
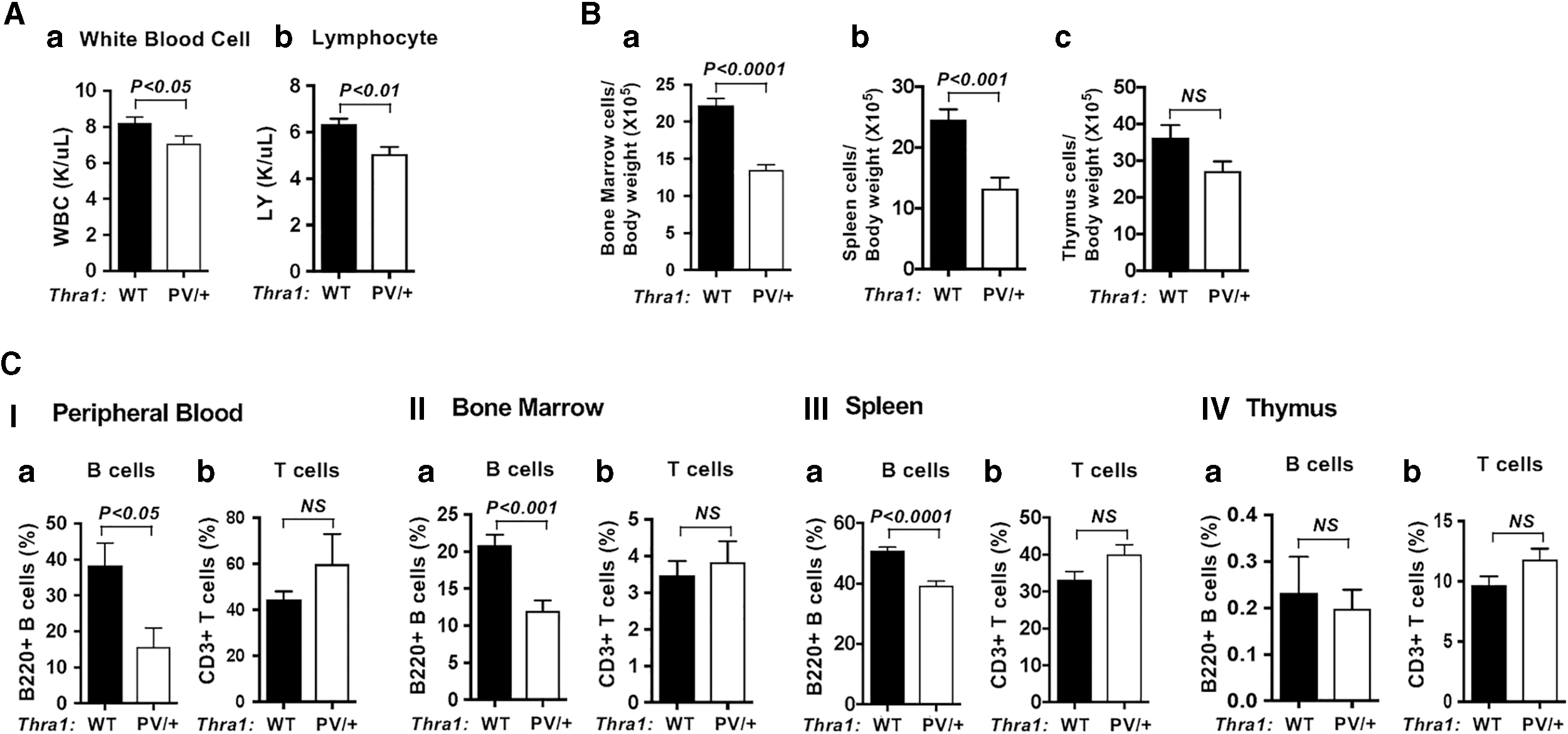
Decreased B lymphocytes in the lymphatic organs of Thra1PV/+
mice. (
We next assessed whether the impaired B lymphopoiesis is limited to TRα1 mutant isoform by comparing the B and T cell numbers in the bone marrow, spleen, and thymus of Thra1PV/+ mice and ThrbPV/+ mice (Supplementary Fig. S3). The ThrbPV/+ mouse harbors the same dominant negative PV mutation in the C-terminal corresponding position of TRβ as in TRα1PV (13). No significant differences in the number of B cells were found in the bone marrow, spleen, and thymus of ThrbPV/+ mice compared with WT mice (Supplementary Fig. S3[I–III]a, bars 3–4). These data indicate that, in contrast to mutations of TRα1, TRβ mutations did not affect B lymphopoiesis and that the regulation of B lymphopoiesis is TRα1-dependent.
Suppressed B cell development in the bone marrow and spleen of Thra1PV/+ mice
The marked decrease in the B cell production shown in the peripheral blood, spleen, and bone marrow of Thra1PV/+ mice prompted us to hypothesize that these decreases resulted from defects in B cell development. As shown in Figure 2A, the development of B cells is a well-orchestrated process initiated from hematopoietic stem cells (HSCs) in the bone marrow. HSCs differentiate into common lymphoid progenitors (CLPs). B cell precursors expressing the B cell lineage marker, B220, can be differentiated from pre/pro-B to immature B cells according to their differential expression of cell surface markers during development in the bone marrow (14,15). As shown in Figure 2B, total B cells (B220+) and B cell precursors, including pre/pro-B (B220+IgM−), pro-B cells (B220+IgM−CD43+), pre-B cells (B220+IgM−CD43−), immature B cells (B220+IgM+), and mature-recirculating B cells (B220highIgM+), were significantly lower in the bone marrow of Thra1PV/+ mice than in the bone marrow of WT mice (Fig. 2B and Supplementary Fig. S4A).
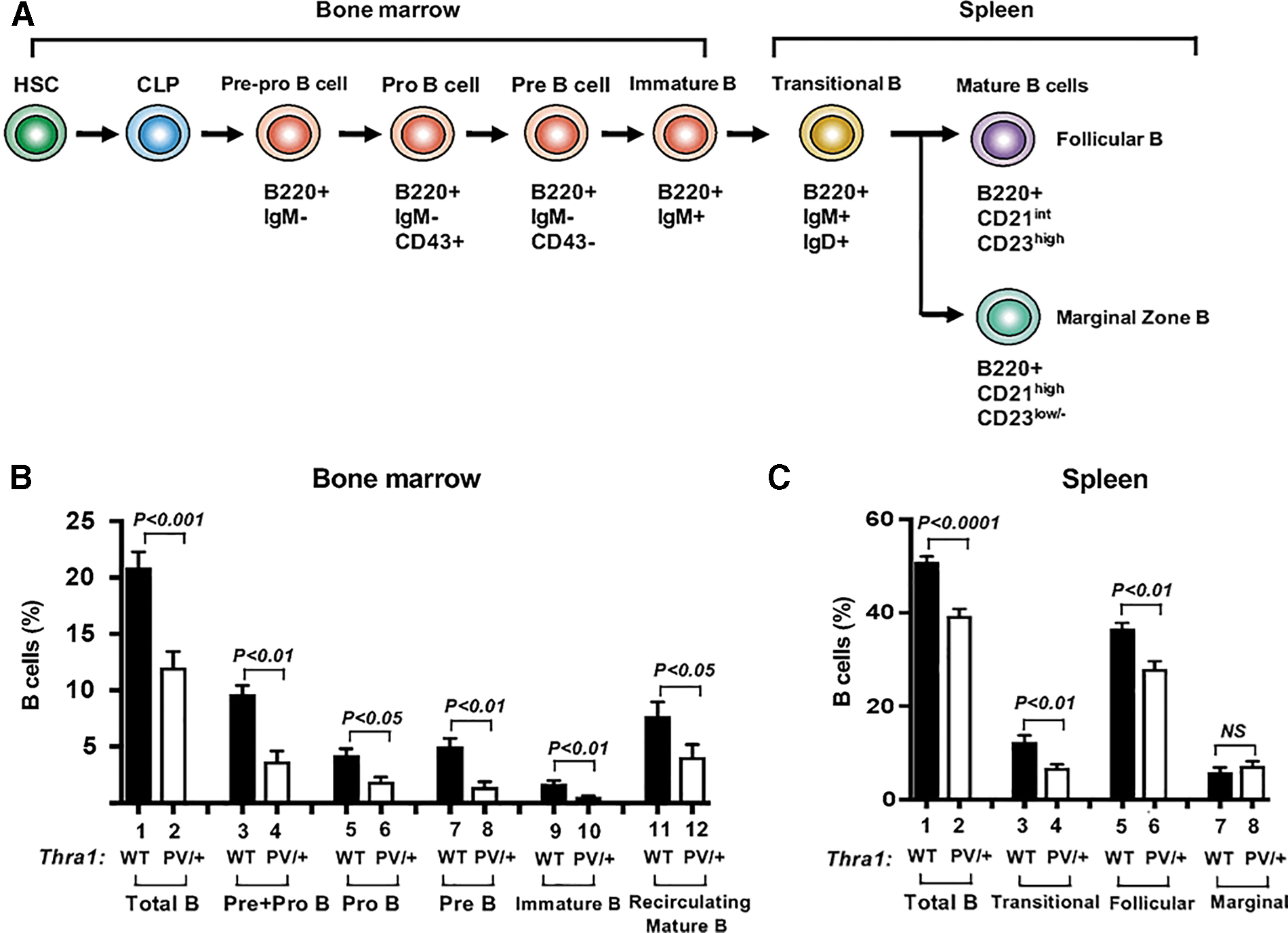
Defective development of B lymphocytes in the bone marrow and spleen of Thra1PV/+
mice. (
The maturation of immature B cells in the bone marrow through transitional stages to mature B cells occurs in the peripheral secondary lymphoid organs, such as the spleen (Fig. 2A). A small population of transitional B cells moves to the marginal zone in the spleen and remains in the spleen as naive noncirculating marginal zone B cells (16). However, most of transitional B cells mature into naive long-lived follicular B cells, which continue circulating to the follicles of the spleen, to the lymph nodes, and to the bone marrow (17). Figure 2C shows that total B cells (B220+) and transitional B cells (B220+IgM+IgD+) were significantly lower in the spleen of Thra1PV/+ mice than in the spleen of WT mice (Fig. 2C, bars 1–4, and Supplementary Fig. S4[B]a and c). In addition, follicular B cells (B220+CD23highCD21int) were significantly lower in the spleen of Thra1PV/+ mice than in the spleen of WT mice (Fig. 2C, bars 5–6). However, no apparent differences in the marginal zone B cells (B220+CD23low/− CD21high) were observed between WT and Thra1PV/+ mice (Fig. 2C, bars 7–8, and Supplementary Fig. S4[B]b and d). Taken together, these data indicate that TRα1PV mutation impaired early B cell development in the bone marrow (pre/pro-B cells differentiation initiated from CLP) and mature B cells (follicular B cells) in the spleen.
Impaired B cell development is mediated by decreased expression of Ebf1, Tcf3, and Pax5 in the bone marrow and spleen of Thra1PV/+ mice
EBF1 (Early B cell Factor 1), TCF3 (Transcription Factor 3, E2A), and PAX5 (Paired box 5) are necessary for B cell commitment and lineage specification (18 –21). In the absence of these critical regulators, B cell development is aborted at the earliest stage (22,23). Loss of PAX5 redirects B cells into other lineages (Fig. 3A) (24). We found that the expressions of the Ebf1, Tcf3, and Pax5 genes were lower by 44%, 44%, and 69%, respectively, in the bone marrow of Thra1PV/+ mice than in the bone marrow of WT controls (Fig. 3B[I]a–c). The expressions of the Ebf1, Tcf3, and Pax5 genes were 65%, 70%, and 72% lower, respectively, in the spleen of Thra1PV/+ mice than in the spleen of WT mice (Fig. 3C[I]a–c). Furthermore, the protein levels of EBF, TCF3, and PAX5 were lower in the bone marrow of Thra1PV/+ mice than in the bone marrow of WT mice (51%, 76%, and 67%, respectively, Fig. 3B[II]i–ii) and in the spleen (62%, 68%, and 73%, respectively; Fig. 3C[II]i–ii) of Thra1PV/+ mice than in the spleen of WT mice. These data suggest that TRα1PV acted at the critical steps of differentiation from CLP to pre/pro-B cells to cause defective B cell maturation development.
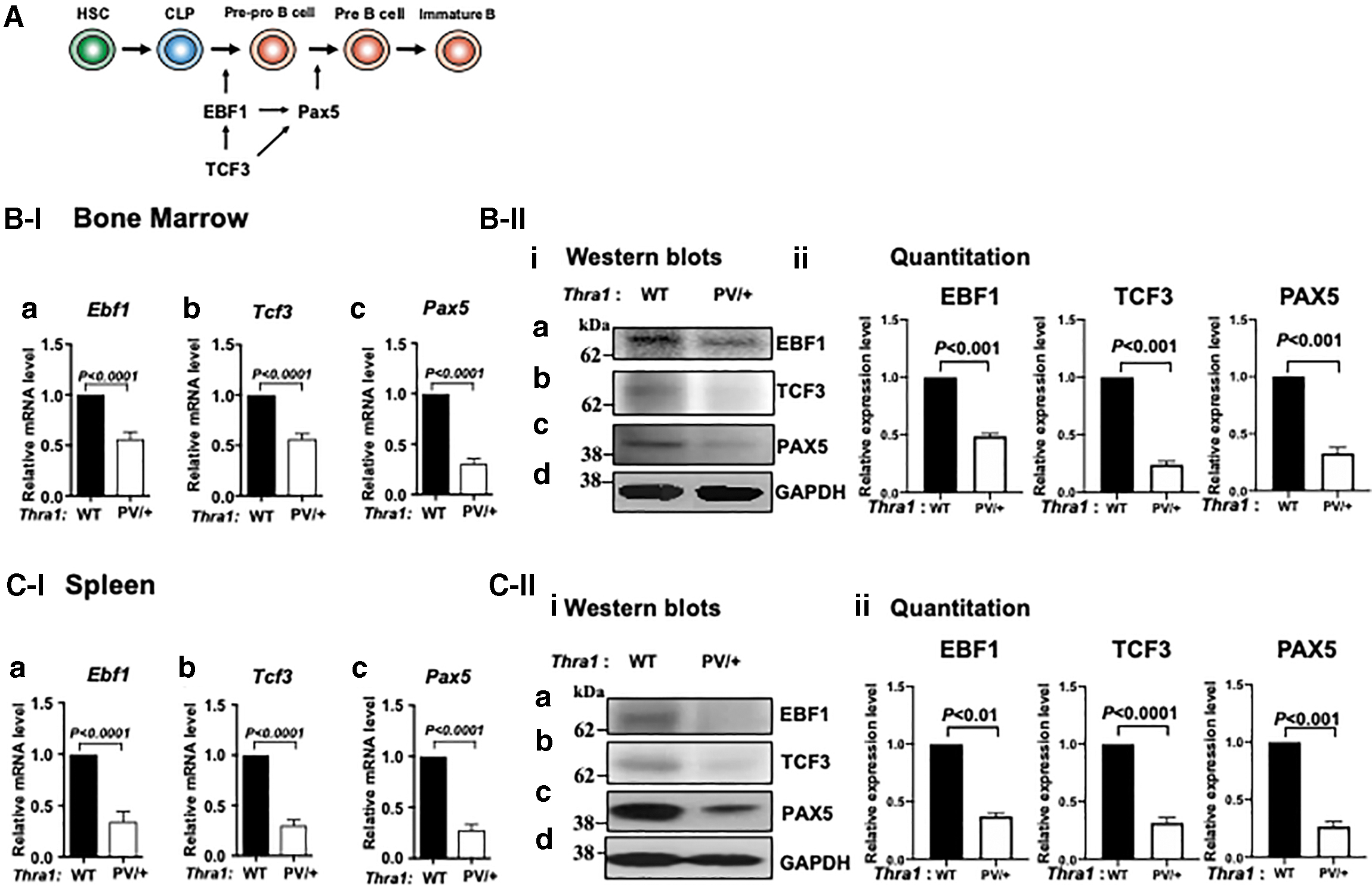
Decreased expression of Ebf1, Tcf3, and Pax5 in the bone marrow and spleen of Thra1PV/+
mice. (
The expression of Ebf1, Tcf3, and Pax5 is regulated by T3 in Thra1PV/+ mice
We next ascertained whether T3 regulated the expression of three critical genes—Ebf1, Tcf3, and Pax5—by rendering WT and Thra1PV/+ mice either hypothyroid via treatment with PTU diet or hyperthyroid via T3 treatment in those PTU-treated mice. In the bone marrow of WT mice, the mRNA expression of the Ebf1, Tcf3, and Pax5 was significantly lower in hypothyroid mice than in untreated mice (Fig. 4a–c, bar 3 vs. 1). The expression of the Tcf3, Ebf1, and Pax5 was significantly greater in the bone marrow of hyperthyroid WT mice than in the bone marrow of hypothyroid WT mice (Fig. 4a–c, bar 5 vs. 3). The expression of Ebf1, Tcf3, and Pax5 mRNA was consistently lower in the bone marrow of untreated Thra1PV/+ mice than in the bone marrow of WT mice (Fig. 4a–c, bar 2 vs. 1). But the expression of Ebf1, Tcf3, and Pax5 mRNA in hyperthyroid Thra1PV/+ mice was not significantly higher than that in hypothyroid Thra1PV/+ mice (Fig. 4a–c, bar 6 vs. 4). These results indicated that TRα1PV had lost T3 binding activity and could not regulate the expression of these three key regulators. Taken together, these results showed that mutations in TRα1 (such as TRα1PV) suppressed the expression of Tcf3, Ebf1, and Pax5 genes to impair B lymphopoiesis in Thra1PV/+ mice. These results indicate that Ebf1, Tcf3, and Pax5 genes were positively regulated by T3 mediated by TRα1 in the bone marrow.
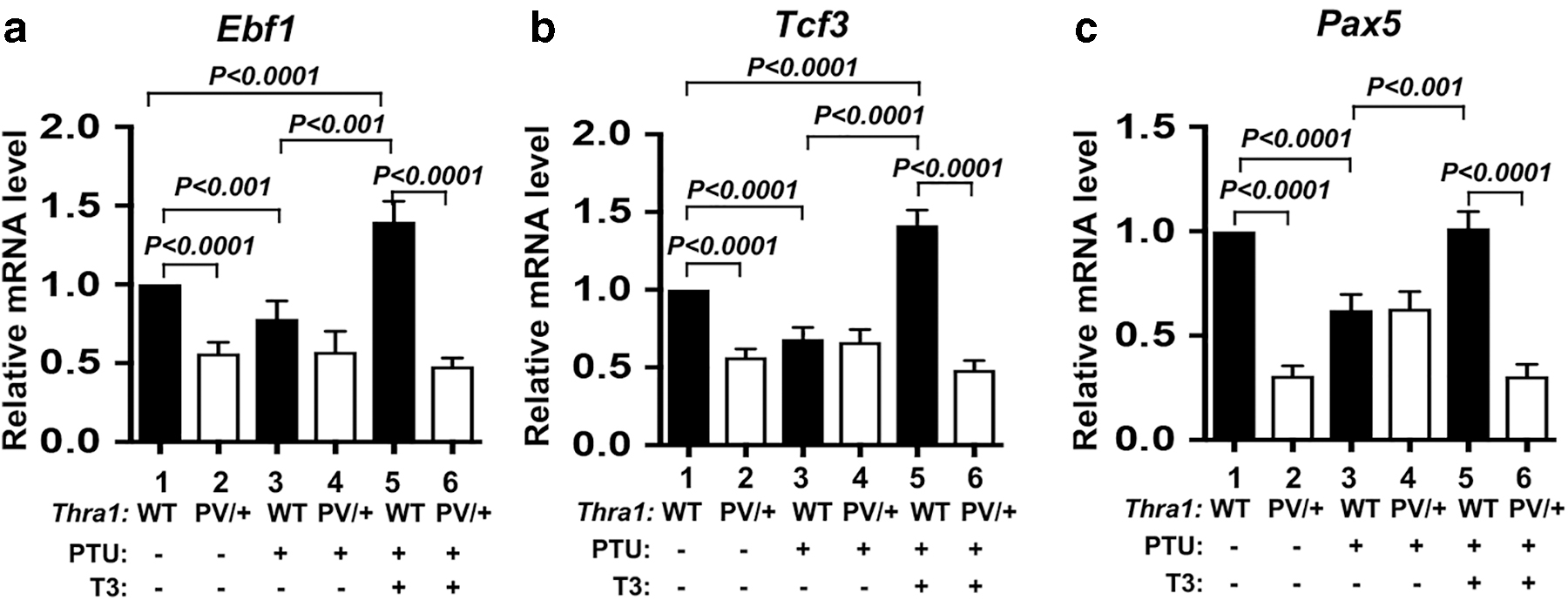
Effect of T3 on the regulation of Ebf1, Tcf3, and Pax5 genes in the bone marrow of Thra1PV/+
mice. Effects of thyroid hormone on the expression of the Ebf1 (
TRα1 directly regulates the expression of the Ebf1 gene in the bone marrow
Because of its critical role in B cell development, we focused on the elucidation of the TRα1 cis-regulatory elements on the promoter of the Ebf1 gene. TRs regulate target gene expression through binding with thyroid hormone response elements (TREs) in the promoters. We used search function in Microsoft Word program to search for consensus sequence of putative TREs [hexa-nucleotide “half-site” (A/G)GGT(C/A/G)A)]. We found putative five half-site TRE sequences in the promoter encompassing the promoter sequences of −536 bp to +299 bp in the Ebf1 gene (Fig. 5A). Using a luciferase construct containing the region of “−536 to +299,” we found that T3-dependent transcriptional activity of TRα1 is mediated by the promoter region of −536 to +299 (compare bar 4 with bar 3, Fig. 5B[a]). We constructed a luciferase reporter to determine if the three putative TREs in Ebf1 #1, (encompassing −492 to −291) could mediate T3-dependent transcriptional activity of TRα1. However, no significant T3-dependent transcriptional activity of TRα1 was detected in the luciferase reporter containing Ebf1 #1 (Fig. 5B[b]). However, the luciferase reporter we constructed to assess the two putative TREs in Ebf1 #2 (encompassing −155 to +83) showed twofold T3-dependent transcriptional activity of TRα1 (compare bar 4 with bar 3, Fig. 5B[c]). To further validate that Ebf1 #2 was the TRα1 binding site, we carried out ChIP assays by using monoclonal antibody C4, which recognizes the C-terminal sequences of WT TRα1 (10). A significant binding of WT TRα1 to Ebf1 #2 was detected (Fig. 5B[d]), but less binding to Ebf1 #2 was detected in the bone marrow cell of heterozygous Thrα1PV/+ mice (Fig. 5B[d]).
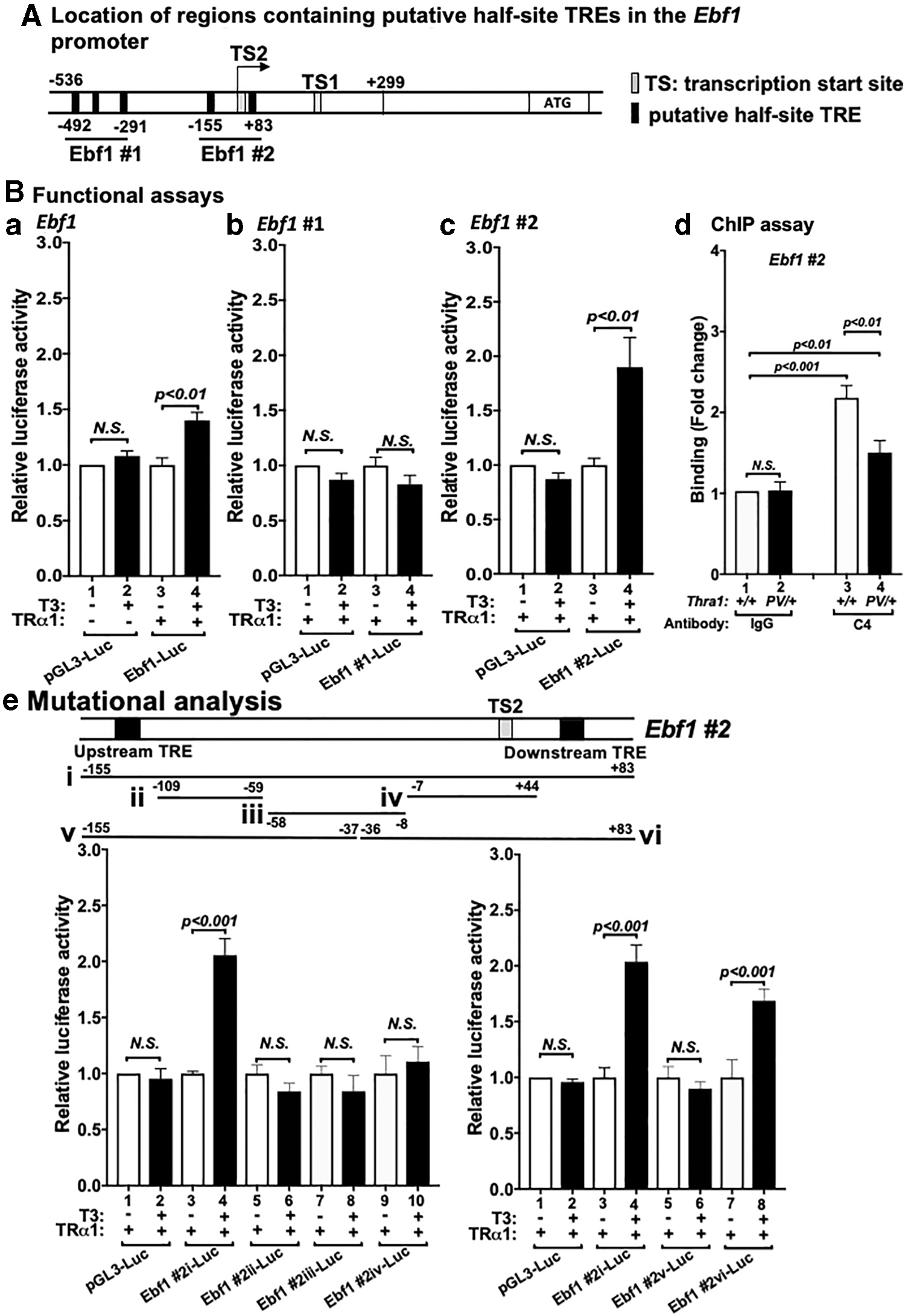
Identification of TREs in the Ebf1 promoter. (
We further prepared three truncated luciferase reporter constructs that did not contain either of the half-site TRE and found that no T3-dependent transcriptional activity of TRα1 was observed (Fig. 5B[e]ii, iii, and iv). Two additional luciferase reporters containing only one of the two individual TREs as shown in Figure 5B[e]v (upstream TRE, Fig. 5B[e]) and Figure 5B[e]vi (downstream TRE) showed that T3-dependent transcription activity of TRα1 was only detected in the luciferase reporter containing the “downstream” TRE (bar 8 vs. 7 in the right panel data graph of Fig. 5B[e]). Taken all together, these data indicate that we have identified one TRE to interact with T3-bound TRα1 to directly regulate the expression of the Ebf1 gene to impair lymphopoiesis.
Discussion
In the present study, we found a significant reduction of B lymphocytes in the peripheral circulation, bone marrow, and spleen. The expression of key regulators of B cell development, such as Tcf3, Ebf1, and Pax5, was significantly decreased in the bone marrow and spleen of Thra1PV/+ mice. We further provided direct evidence to show that the Ebf1 gene was a TRα1/T3 directly regulated gene. Thus, mutations of TRα1 could impair B cell development in the bone marrow via suppression of key regulators in B lymphopoiesis. These provide new insights into understanding B lymphopoiesis in RTHα patients. The discovery that mutations of the THRA gene could impact B cell development, as shown in the present study, should provide a strong rationale to analyze the B cell abundance in lymphoid tissues of RTHα patients when it is feasible.
The effect of thyroid hormone on B lymphopoiesis has been previously reported. Earlier studies using the mouse strain, Snell dwarf (dw/dw), deficient in growth hormone (GH), prolactin (PRL), insuline-like growth factor-1 (IGF-1), thyrotropin, T3, and thyroxine (T4) due to mutation in the pit-1 transcription factor, showed defective B lymphopoiesis. However, only treatment of Snell dwarf with levothyroxine (LT4), but not GH, IGF-1, or PRL, restored the frequency of B cell lineage cells to normal and to increase marrow cellularity (25). These results indicated an obligatory role of thyroid hormones in the B cell development. This conclusion was further supported by additional analysis of the thyroid hormone-deficient hypothyroid (hyt/hyt) mice (26,27). Hypothyroid (hyt/hyt) mice showed a deficiency of CD45R+IgM− B progenitors, which was reversed by treatment of the mice with T4 (28). These results demonstrated the obligatory role of thyroid hormone in the B cell lineage development. Recently, analysis of TRα1/TRβ double knockout mice further confirmed that it was TRα1, but not TRβ, that was involved in the regulation of B cell development (29), which is in line with our findings, showing that mutations of TRα1 as in Thra1PV/+ mice impaired B cell development (Supplementary Fig. S3).
Previous studies demonstrated that the effect of thyroid hormone on lymphopoiesis is specific to B cell lineage (29). Consistent with these earlier studies, we found that Thrα1PV/+ markedly decreased pre-B cells. Furthermore, we also found that the inhibitory action of Thrα1PV/+ was initiated further upstream of pre-B cells. Pre/pro-B cells was immediately downstream of CLP, which was also markedly decreased in the bone marrow of Thra1PV/+ mice. CLP progenitors differentiate to pre/pro-B cells. Differentiation from CLP to pre/pro-B cells is known to be regulated by critical transcription factors, EBF and E2A (20). The expression of these two critical regulators was suppressed in the bone marrow and spleen of Thra1PV/+ mice. We also identified TREs on the promoter region of the Ebf1 gene (Fig. 5). Therefore, our studies have provided a molecular basis to show how TRα1 mutations could cause defective B lymphopoiesis. Importantly, the present study provides rationales for further investigations of B cell development in RTHα patients beyond determination of peripheral white blood cells.
Footnotes
Acknowledgment
We thank Dr. Mikael Sigvardsson (Lude University, Sweden) for the gift of ebf1 promoter plasmids.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3