
Abstract
Background:
Thyroxine (T4) to triiodothyronine (T3) deiodination in the hypothalamus/pituitary is mediated by deiodinase type-2 (D2) activity. Dio2(−/−) mice show central resistance to exogenous T4. Patients with resistance to exogenous thyroxine (RETH) have not been described. The aim of this study was to identify hypothyroid patients with thyrotropin (TSH) unresponsiveness to levothyroxine (LT4) and to characterize the clinical, hormonal, and genetic features of human RETH.
Methods:
We investigated hypothyroid patients with elevated TSH under LT4 treatment at doses leading to clinical and/or biochemical hyperthyroidism. TSH and free T4 (fT4) were determined by chemiluminescence, and total T4, T3, and reverse T3 (rT3) by radioimmunoassay. TSH/fT4 ratio at inclusion and T3/T4, rT3/T4, and T3/rT3 ratios at follow-up were compared with those from patients with resistance to thyroid hormone (RTH) due to thyroid hormone receptor-β (THRB) mutations. DIO2, including the Ala92-D2 polymorphism, selenocysteine binding protein 2 (SECISBP2), and THRB were fully sequenced.
Results:
Eighteen hypothyroid patients (nine of each sex, 3–59 years) treated with LT4 showed elevated TSH (15.5 ± 4.7 mU/L; reference range [RR]: 0.4–4.5), fT4 (20.8 ± 2.4 pM; RR: 9–20.6), and TSH/fT4 ratio (0.74 ± 0.25; RR: 0.03–0.13). Despite increasing LT4 doses from 1.7 ± 1.0 to 2.4 ± 1.7 μg/kg/day, TSH remained elevated (6.9 ± 2.7 mU/L). Due to hyperthyroid symptoms, LT4 doses were reduced, and TSH increased again to 7.9 ± 3.2 mU/L. In the euthyroid/hyperthyrotropinemic state, T3/T4 and T3/rT3 ratios were decreased (9.2 ± 2.4, RR: 11.3–15.3 and 2.5 ± 1.4, RR: 7.5–8.5, respectively) whereas rT3/T4 was increased (0.6 ± 0.2; RR: 0.43–0.49), suggesting reduced T4 to T3 and increased T4 to rT3 conversion. These ratios were serum T4-independent and were not observed in RTH patients. Genetic testing was normal. The Ala92-D2 polymorphism was present in 7 of 18 patients, but the allele dose did not correlate with RETH.
Conclusions:
Human RETH is characterized by iatrogenic thyrotoxicosis and elevated TSH/fT4 ratio. In the euthyroid/hyperthyrotropinemic state, it is confirmed by decreased T3/T4 and T3/rT3 ratios, and elevated rT3/T4 ratio. This phenotype may guide clinicians to consider combined T4+T3 therapy in a targeted fashion. The absence of germline DIO2 mutations suggests that aberrant post-translational D2 modifications in pituitary/hypothalamus or defects in other genes regulating the T4 to T3 conversion pathway could be involved in RETH.
Introduction
Thyroxine (T4
Dio2(−/−) mice show resistance to the feedback effect of T4 on TSH synthesis and secretion by the pituitary, leading to elevated plasma TSH and T4, normal T3, and abnormally decreased T3/T4 ratios in different tissues (6,7). Such a concomitant increase in plasma TSH and T4 in Dio2(−/−) mice partially mimics the hormone phenotype of thyroid hormone receptor beta, Thrb(−/−) mice and of patients with thyroid hormone resistance (RTH) due to mutations in the thyroid hormone receptor-β (THRB) gene (8 –10), the predominant thyroid hormone receptor subtype in the pituitary.
Administration to propylthiouracil to Dio2(−/−) mice caused a further elevation of TSH, which can only be suppressed with L-triiodothyronine (L-T3), but not with levothyroxine (LT4) (6). In accordance, thyroidectomized rats require a combination of LT4 and L-T3 (T4+T3) to normalize thyroid hormone levels in all tissues, stressing the physiological relevance of the T3 secreted from the thyroid gland in the regulation of the HPT axis (11).
The selenocysteine binding protein 2 (Secisbp2) mediates the insertion of selenocysteine in selenoproteins, including iodothyronine deiodinases (12). Secisbp2 knockout (KO) mice exhibit an elevation of plasma TSH and a low T3/T4 ratio, pointing to D2 as the most affected iodothyronine deiodinase in this defect (13). Patients with SECISBP2 mutations have thyroid hormone abnormalities, in addition to a complex array of visual, reproductive, immune, cutaneous, and movement disorders (14,15).
To our knowledge, no pathogenic mutations have been identified in any of the human iodothyronine deiodinase (DIO1, DIO2, DIO3) genes (6,16,17). However, a very common genetic variant in DIO2 (Thr92Ala-DIO2, rs225014; Mean Allelic Frequency: 40% in the general population) was recently associated with a subtle but significant decrease in serum T3 levels after thyroidectomy (18). The DIO2 polymorphism reduced T4 to T3 conversion in cultured cells (18). The impaired T3 generation in Ala92-D2 mutant mice is due to an endoplasmic reticulum to Golgi transition defect (19). Ala92-D2 rodents show signs of brain hypothyroidism (19). Interestingly, the administration of T4+T3 to these mice, but not T4 alone, rescued a behavioral phenotype of increased anxiety and depression (20).
Many hypothyroid patients treated with LT4 alone manifest persisting symptoms of hypothyroidism and psychological ill-being (21). However, current international guidelines recommend LT4 therapy alone as the standard treatment of hypothyroidism. A recent meta-analysis of 11 randomized clinical trials compared T4 alone and T4+T3 therapies for hypothyroidism and found no better long-term outcomes for the combined treatment (21). The American Thyroid Association guidelines found insufficient evidence to support the routine use of T4+T3 therapy in primary hypothyroidism except for patients feeling unwell on LT4 alone (22). The European Thyroid Association recommends the combination therapy only in adult patients undergoing thyroidectomy, and subsequent LT4 treatment, who still present with symptoms of hypothyroidism and plasma-free T3 (fT3) levels lower than preoperative values (23).
Although T4+T3 therapy in human hypothyroidism remains controversial, it is plausible that some patients with persistent complaints may benefit from this therapy. In most clinical trials, the assignment of patients to T4 or T4+T3 therapies was random, and the analysis did not include genotypic or specific thyroid function test profiles (21,24 –26). It has been recently reported that patients harboring two common polymorphisms, in MCT10 (rs17606253) and DIO2 (rs225014, Ala92-DIO2), preferred T4 and T3 combination therapy (27). Besides the relevance of these polymorphisms in the treatment of hypothyroidism, a clinical condition such as resistance to LT4 has not been defined. Identification of this condition through sensitive biomarkers would enable a targeted selection of specific hypothyroid patients for combined T4+T3 therapy.
In this study, we aimed at the identification of hypothyroid patients who persistently presented with uncontrollable TSH levels while receiving LT4 at doses causing clinical and/or biochemical hyperthyroidism. The clinical presentation and responses to LT4 were consistent with pituitary resistance to exogenous thyroxine (RETH).
Patients and Methods
Patient selection and protocol
Patients with clinical suspicion of RETH were recruited for the study if they met the following inclusion criteria: LT4 treatment for a primary thyroid disorder. TSH concentration (>5 mIU/L) and TSH/fT4 ratio (>0.15 mIU/pmol) persistently high despite the use of regular LT4 doses (1–4 μg/kg/day for children; 1–2 μg/kg/day for adolescents and adults). TSH and TSH/fT4 normalized only by using LT4 doses causing clinical or biochemical hyperthyroidism (fT4 > 20 pM).
Exclusion criteria:
Irregular or noncompliance with LT4 treatment.
Suspected or diagnosed malabsorption disorders (e.g., celiac disease).
Treatment with drugs interfering with thyroid function or metabolism (e.g., ferrous sulfate, amiodarone, growth hormone, estrogens).
Children with congenital hypothyroidism younger than 1 year of age.
After inclusion, clinicians were instructed to progressively reduce LT4 to attain clinical euthyroidism, or to return fT4 to the normal range, even at the expense of TSH elevation. Under euthyroidism (normal fT4), thyroid function tests were performed and informed consent for genetic studies obtained from index patients and/or their parents or guardians, according to protocols approved by the Ethical Committee of La Paz University Hospital, Madrid, Spain (CEIC-PI1496).
Serum thyroid function tests
Fasting blood samples were collected before LT4 administration. Serum TSH, fT4, anti-thyroperoxidase (anti-TPO), and anti-thyroglobulin (anti-TG) antibodies were determined by chemiluminescent immunometric assay (Access Immunoassay Systems 2006; Beckman Coulter, Milan, Italy). T3, T4, and rT3 were measured by radioimmunoassay (Beckman Coulter, Immunotech, Czech Republic, and ZenTech, Belgium, respectively). Normal ranges from the manufacturer's protocols (TSH: 0.4–4.5 mIU/L; fT4: 11.6–20.6 pM; T3: 1.2–2.8 nM; T4: 69–160 nM; rT3: 0.14–0.54 nM) were used and confirmed by serum samples from healthy donors. Normal ranges for TSH/fT4 (0.03–0.13), T3/T4 (11.3–15.3; calculated as T3 nM/T4 nM × 103), rT3/T4 (0.43–0.49; calculated as rT3 nM/T4 nM × 102), and T3/rT3 (7.5–8.4) ratios were calculated and evaluated from available ranges (28 –33).
Total cholesterol and low-density lipoprotein (LDL) cholesterol were determined by enzymatic colorimetry assay (COBAS c702 Autoanalyzer; Roche Diagnostic, Switzerland). The sex hormone-binding globulin (SHBG) was determined by electrochemiluminescence immunoassay (COBAS e801 Autoanalyzer; Roche Diagnostics, Germany). Normal ranges from the manufacturers were used. For comparison, all analytes were also determined in five patients with classical RTH, of whom three had the THRB mutations p.R243Q, p.P453S, and p.R438H, respectively, and two had p.L450F.
DNA isolation and genotyping
Genomic DNA was isolated from white blood cells by using the Chemagic Magnetic Separation Module I (Chemagic MSM I, Chemagen; PerkinElmer, Germany), according to the manufacturer's instructions. The complete coding regions of DIO2, SECISBP2, and THRB (including the β2-specific exon) were screened for mutations by using PCR and Sanger sequencing on an automated DNA sequencer (3100 Genetic Analyzer; Applied Biosystems, CA) (34 –36). The 54-basepair region defining the SECIS (Selenocysteine Insertion Sequence) element at the 3′-untranslated region (UTR) of DIO2 was also genotyped (37,38). Primer sequences are provided in Supplementary Table S1.
Statistical analyses
Statistical analysis was performed by using GraphPad Prism software version 5 (San Diego, CA). Hormonal profiles and descriptive data were reported as mean ± standard deviation. The paired t-test was used to analyze normally distributed data. Differences between means were assessed with the Student two-tailed t-test for independent samples and considered significant when p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Correlation analyses were performed by using the Pearson Correlation test and considered strong (r > 0.6), moderate (r = 0.3–0.6), or weak (r < 0.3) for direct or inverse correlations.
Results
Study cohort with RETH
The study cohort was composed of 18 patients, 9 females and 9 males, with ages ranging from 3 to 59 years with clinical suspicion of RETH (Table 1). Four patients were excluded: one female with thyroiditis and Turner Syndrome under growth-hormone treatment, a male with thyroiditis and positive anti-gliadin antibodies under investigation for celiac disease, and two adolescents with Hashimoto's thyroiditis and inconsistent adherence to LT4 treatment.
Clinical and Hormonal Characteristics of Patients at Suspicion and Confirmation of Resistance to Exogenous Thyroxine
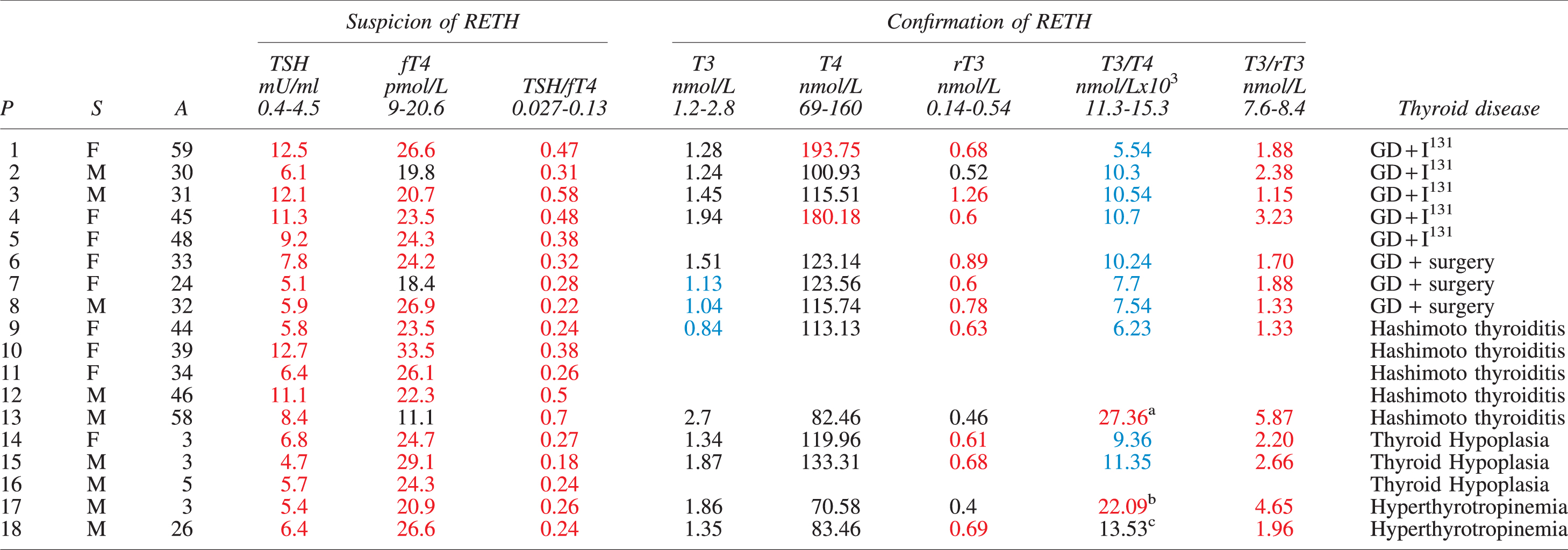
P13 was under T4+T3 substitution therapy.
P17 has a thyroid gland in situ (intact T3 secretion from thyroid).
P18 has a thyroid gland in situ (intact T3 secretion from thyroid). He had withdrawn LT4 for intolerance 3 months before the confirmation phase.
P, S, A, TSH, fT4, and TSH/fT4 ratios are presented at suspicion of RETH (iatrogenic hyperthyroid phase). Total T3 (T3), total T4 (T4), rT3, T3/T4 ratios, and T3/rT3 ratios were determined for confirmation of RETH (euthyroid phase). Thyroid diseases leading to hypothyroidism and therapeutic measures are indicated: GD+ I131, Graves' disease followed by radioactive ablation with I131; GD+surgery, Graves' disease treated by total or near total thyroidectomy. Normal ranges for TSH and fT4 are specified in the Methods section. TSH/fT4 ratio is t from Bjørn et al. (28). Normal ranges for T3, T4 and rT3 are specified in the Methods section. Normal ranges for T3/T4 and T3/rT3 ratios are from Gullo et al. (29); Oto et al. (30); Strich et al. (31); Ruhla et al. (32); Chopra et al. (33) and were used based on the age of patients. Values below (blue), above (red) and within (black) normal ranges are indicated.
A, age; fT4, free T4; GD, Graves disease; LT4, levothyroxine; P, patients; RETH, resistance to exogenous thyroxine; rT3, reverse T3; S, sex; T3, triiodothyronine; T4, thyroxine; TSH, thyrotropin.
Color images are available online.
Patients entering the study received LT4 for different primary thyroid disorders including Graves-Basedow's disease (n = 8) requiring 131-radioiodine thyroid ablation (n = 5) or total thyroidectomy (n = 3), autoimmune hypothyroidism (Hashimoto's disease, n = 5), congenital severe thyroid hypoplasia (n = 3), and isolated hyperthyrotropinemia with normally sized and located thyroid (n = 2; Table 1).
All patients showed elevated TSH (15.4 ± 4.8 mU/L; reference range [RR]: 0.4–4.5) while receiving LT4 at an initial mean dose of 1.7 ± 1.0 μg/kg/day (Fig. 1A), ranging from 1.3 ± 0.7 μg/kg/day in adults to 2.9 ± 1.1 μg/kg/day in children (22). To normalize TSH, LT4 had been progressively increased reaching a mean dose of 2.38 ± 1.7 μg/kg/day (1.84 ± 0.83 μg/kg/day for adults; 4.2 ± 2.7 μg/kg/day for children), representing increases of 34% and 43%, respectively (p > 0.05). Despite increasing LT4 doses, the TSH remained elevated (6.9 ± 2.7 mU/L; RR: 0.4–4.5). FT4 was elevated or borderline-high (24.3 ± 3.5 pM; RR: 11.6–20.6), and the TSH/fT4 ratio remained strongly elevated (0.36 ± 0.17; RR: 0.03–0.13; Fig. 1; Table 1) (28).
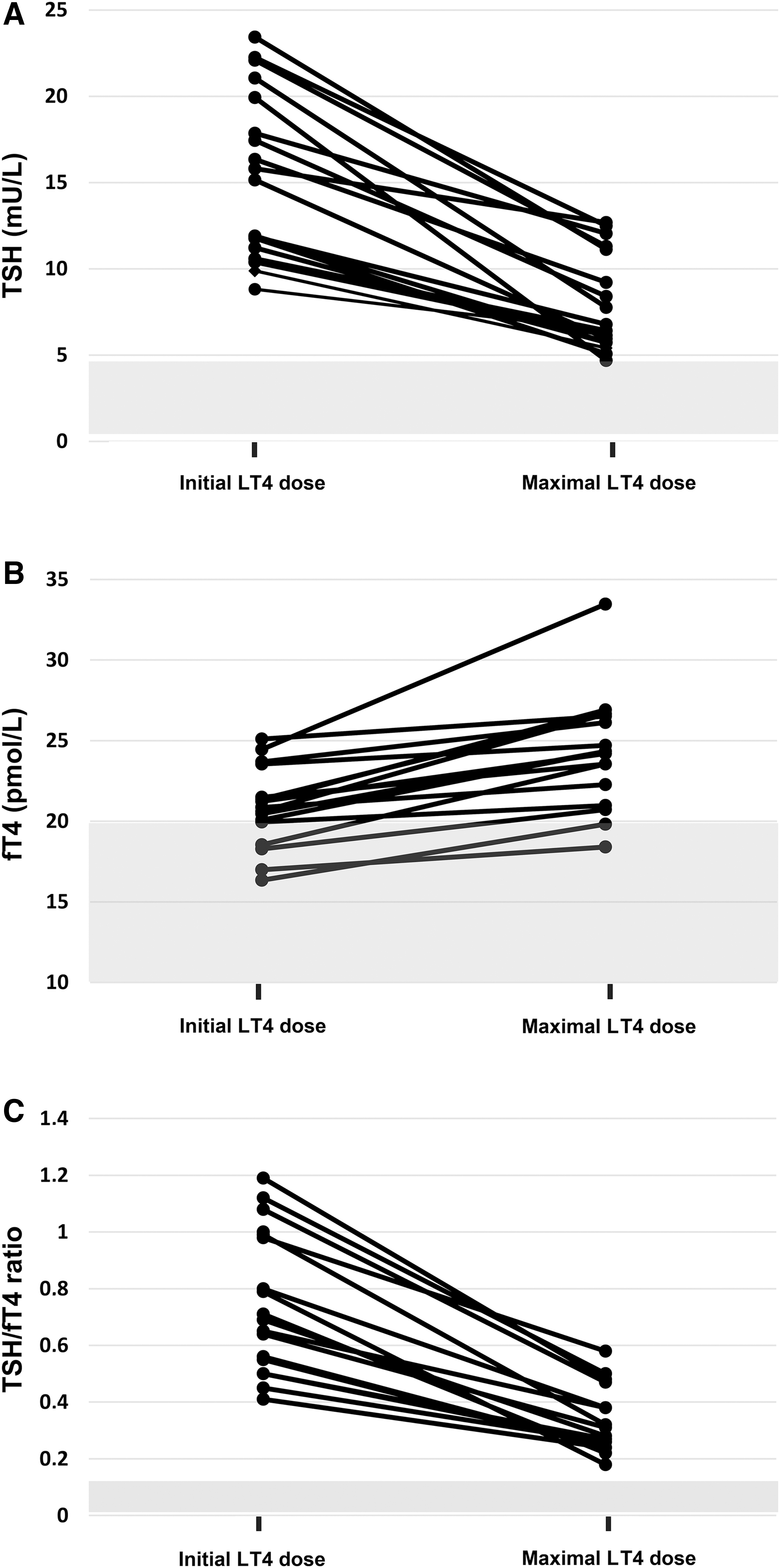
Dynamics of thyroid function in patients at clinical suspicion of RETH. Individual patient's TSH (
Clinical hyperthyroidism was overtly present in five adults (P1, P2, P3, P13, P18). and three children (P14, P15, P17), who showed signs and symptoms such as nervousness, tachycardia, diarrhea, or insomnia. They all had elevated fT4 levels, at the borderline of the hyperthyroid range, except one (P13) who showed normal serum fT4 but elevated TSH/fT4 ratio (Table 1). Two patients (P17, P18) presented with clinical and biochemical hyperthyroidism under moderate-low doses of LT4 for their age (1.4 and 0.7 μg/kg/day, respectively), the lowest in the cohort (Table 1). Six of the adult patients (P1, P3, P4, P6, P7, P18) presented with psychiatric symptoms such as anxiety (n = 1), hyperactivity (n = 3), depression (n = 2), and psychosis (n = 1). These disorders required medication and in one patient led to legal psychiatric incapacitation.
Individual plots of TSH and fT4 indicated abnormal pituitary-thyroid axis set-points (39), suggesting a defect in the central regulation of TSH secretion by thyroid hormones (Supplementary Fig. S1). In this setting, RETH was suspected and LT4 was reduced progressively until clinical or biochemical hyperthyroidism resolved. In the euthyroid state, patients were referred for a centralized thyroid hormone profile and genetic analysis. Follow-up time from the diagnosis of hypothyroidism to referral for suspicion of RETH was variable (14.1 ± 7.2 months).
Metabolism of thyroid hormones and peripheral markers of T4 action
When LT4 doses were reduced (at the expense of TSH elevation), the patients achieved clinical euthyroidism and T4 levels fell within the normal range in most cases (114.6 ± 31.5 nM; RR: 69–160), with individual variations perhaps reflecting the different LT4 doses used (Fig. 2A). T3 decreased to the lower half of the RR (1.4 ± 0.3 nM; RR: 1.2–2.8) (Fig. 2B), and rT3 was elevated in most patients (0.7 ± 0.2 nM; RR: 0.14–0.54) (Fig. 2C). Accordingly, the T3/T4 ratio was decreased (9.2 ± 2.5; RR: 11.3–15.3) (Fig. 2D) (29 –31), the rT3/T4 ratio increased (0.6 ± 0.2; RR: 0.43–0.49) (Fig. 2E) (32), and the T3/rT3 ratio was severely decreased (2.5 ± 1.4; RR: 7.5–8.5) (Fig. 2F) (33). These results are consistent with an impaired capacity of T4 to T3 conversion in RETH patients and an apparently normal capacity for T4 to rT3 inactivation. As rT3 is also a D2 substrate (6,40), increased rT3/T4 ratio is explained by a D2 defect. However, since rT3 is a better substrate than T4 for D1 (the main mechanism for rT3 clearance) (2,16), a defect in D1 cannot be completely excluded. In either case, the T3/rT3 ratio appears the most sensitive marker for clinical diagnosis of RETH, regardless of individual serum T4 levels.
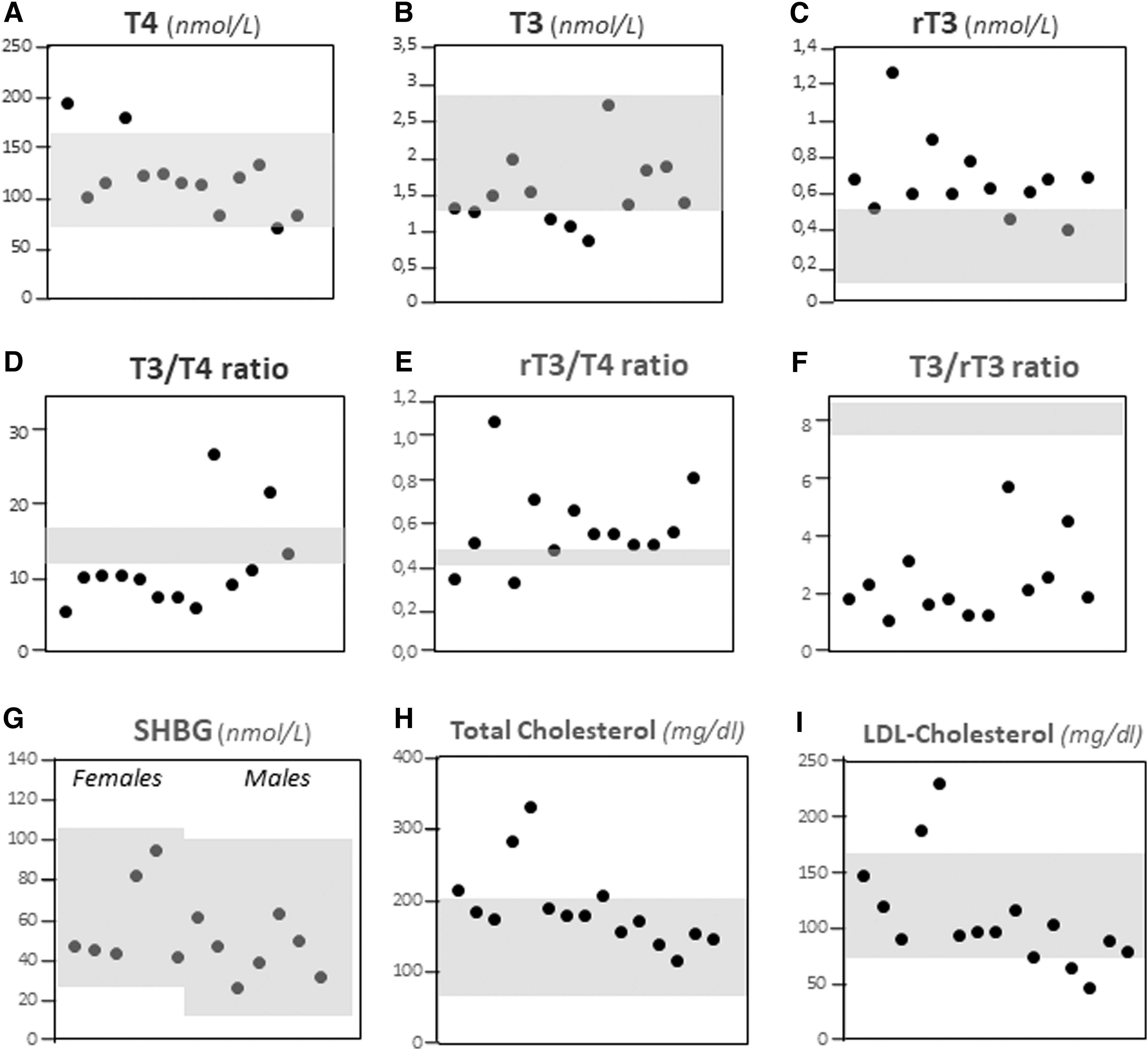
Biochemical markers at diagnosis of RETH. (
T4 to T3 conversion is mainly performed not only by D2 in target organs such as the brain, pituitary, skeletal muscle, heart, and thyroid but also by D1 in other organs such as the liver and kidney. We further investigated two parameters related to TH action in the liver: lipid metabolism and the levels of the SHBG (41 –43). Total and/or LDL cholesterol was elevated in 7 of 14 patients, 5 of whom were on statin therapy (Fig. 3G, H). SHBG was normal in all cases (Fig. 2I). Overall, the data suggest that RETH has more profound effects on the central (hypothalamic/pituitary) than on the peripheral (liver) metabolism of thyroid hormones.
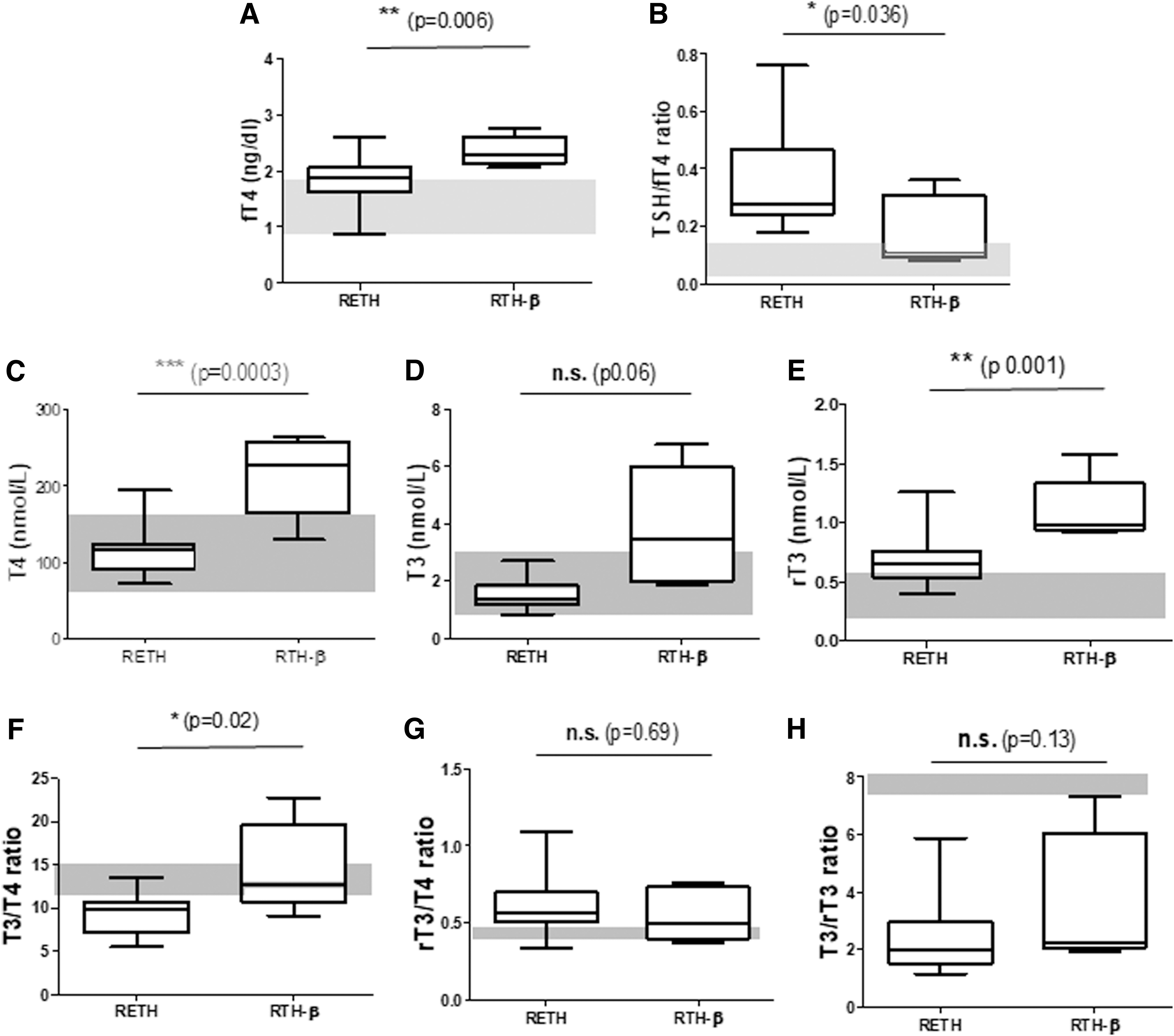
Comparison of thyroid hormone profiles between RETH and RTH-β patients. (
Comparison of thyroid hormone profiles in RETH and RTH-β patients
RETH and the classical RTH caused by THRB gene mutations may share the concomitant increase of TSH and T4. Therefore, we compared the thyroid hormone profile of patients with these entities for discriminative features. At clinical suspicion, serum fT4 levels are elevated in both groups; however, biochemical hyperthyroidism were higher in RTH-β patients (p < 0.006; Fig. 3A). RETH patients had significantly higher TSH/fT4 ratios than RTH-β patients (Fig. 3B). After LT4 dose reduction, T4 can be normalized in RETH patients, suggesting that the euthyroid-hyperthyrotropinemic phase of RETH is the optimal period for discrimination between these entities (p < 0.0003; Fig. 3C). Similarly, T3 is normal in RETH and elevated in RTH-β, with borderline significance (Fig. 3D). Finally, rT3 levels were elevated in both disorders but were significantly higher in RTH-β than in RETH patients (Fig. 3E).
The T3/T4 ratio was normal in RTH-β but significantly reduced in RETH (p < 0.02; Fig. 3E), suggesting a specific defect of T4 deiodination in RETH, and not in RTH-β patients. In contrast, the rT3/T4 ratio was similar in both entities (Fig. 3F), suggesting that T4 degradation pathways are intact in both disorders. The T3/rT3 ratio was similarly reduced in RETH and RTH-β patients (Fig. 3G) for seemingly different reasons: impaired T3 generation in the case of RETH (suggesting defective D2 activity) and increased formation of rT3 in RTH-β as a consequence of the hyperthyroid state. Overall, the results point to a D2 functional deficiency underlying RETH, which is not present in RTH-β.
We also investigated the possible influence of the different T4 levels between RETH and RTH-β patients on the end-result of T4 activation (conversion to T3) and inactivation (conversion to rT3) pathways. The corresponding T3/T4, rT3/T4, and T3/rT3 ratios were correlated to serum T4 concentrations in each group (Fig. 4). In RTH-β patients, serum T4 correlated directly with the T3/T4 ratio (r = 0.59; p = 0.29) (Fig. 4A). In contrast, T4 concentrations correlated inversely with T3/T4 ratio in RETH patients (r = −0.61; p = 0.02) (Fig. 4B). This indicates diminished T4 activation in RETH, which is independent of T4 levels and cannot be overcome by increasing the LT4 dose (Fig. 4B). Interestingly, for the T4 inactivation pathway as a function of T4 levels (Fig. 4C, D), both inversely correlated in RTH-β (r = −0.75; p = 0.014) and RETH (r = −0.49; p = 0.08) patients, suggesting normal dynamics in T4 to rT3 deiodination in both disorders. Finally, the correlation between T4 levels and the T3/rT3 ratios was positive in RTH-β (r = 0.68; p = 0.21 (Fig. 4E) and negative in RETH (r = −0.33; p = 0.27) patients (Fig. 4F), although statistically not significant, suggesting an intrinsic and specific defect of the T4 activation pathway in RETH that may be independent from the substrate concentration.
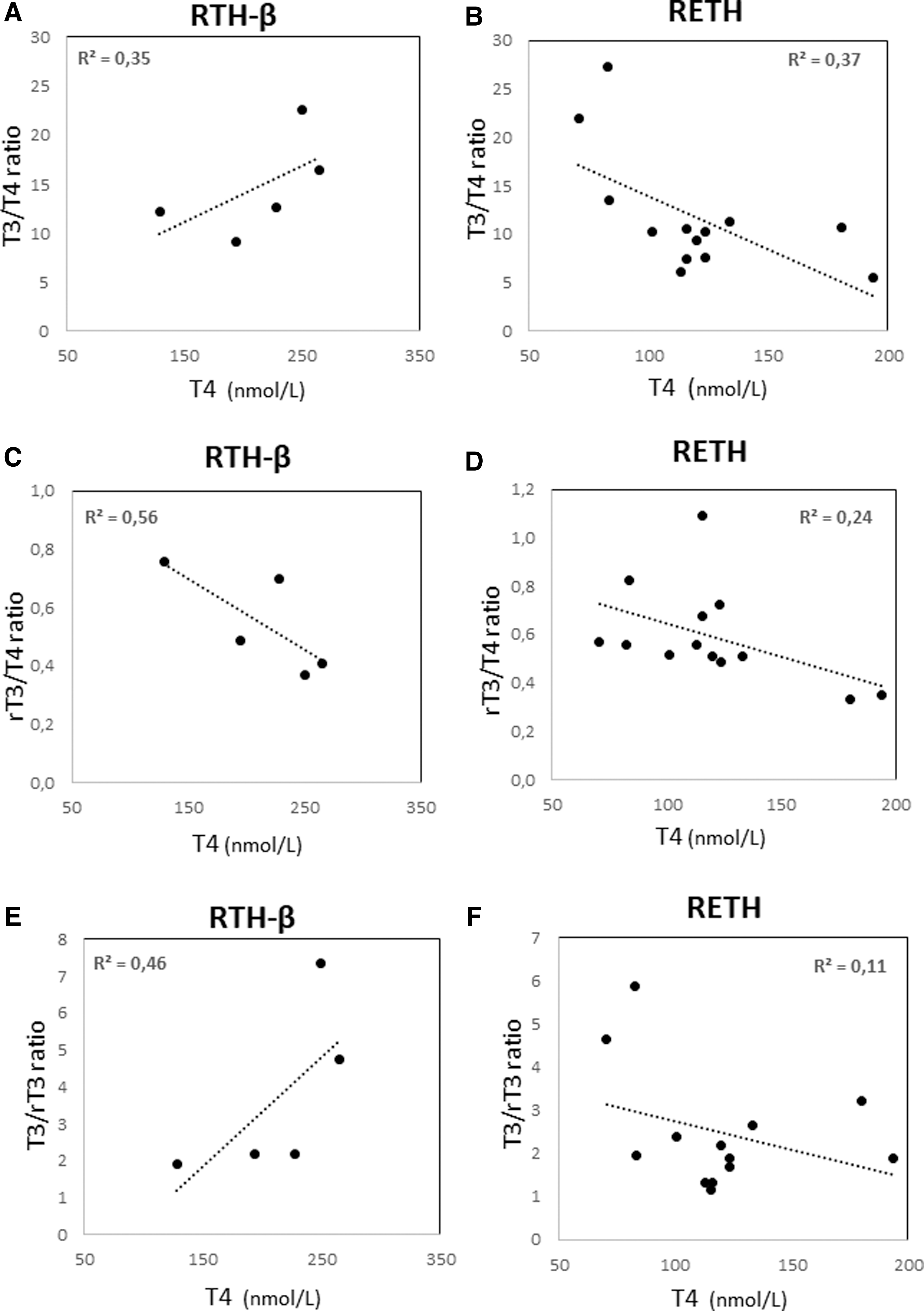
Correlations between serum T4 and thyroid hormone ratios in RTH-β and RETH patients. (
Genetic study of THRB, SECISBP2, and DIO2 coding regions
The results cited earlier strongly suggested deficient T4 activation in RETH patients. Consequently, DIO2, SECISBP2, and THRB genes were fully sequenced in RETH patients. No mutations were identified either in the respective coding regions or in the 3′-UTR of DIO2 coding for the selenocysteine insertion sequence (SECIS element). The Ala92-D2 polymorphism was present in seven patients (five heterozygous and two homozygous) and its allele dosage did not correlate with either TSH/T4 or T3/T4 ratios (Supplementary Fig. 2A, B). The presence of the Ala92-D2 polymorphism in 39% of the RETH cohort (the Minor Allele Frequency of this polymorphism being 40% in Europeans, 1000 Genomes Project Phase 3-) suggests that the Ala92-D2 polymorphism is not the driving cause of the severe phenotype of LT4 resistance characterized here.
Finally, the segregation of RETH in families was rare. The clinical expression of RETH has the requisite of LT4 usage, but the thyroid disorders causing hypothyroidism in our series (autoimmune thyroiditis, thyroidectomy for Graves's disease, thyroid dysgenesis) do not follow Mendelian inheritance, which may strongly compromise the identification of case-clustering of RETH in families.
Response to combined T4+T3 treatment and hypersensitivity to LT4 in individual patients
All patients in the cohort showed a homogeneous hormone pattern at clinical suspicion of RETH, with the hallmark of a largely elevated TSH/fT4 ratio (Table 1). Similarly, after lowering LT4 doses to avoid hyperthyroidism (and assuming the subsequent hyperthyrotropinemia), all patients were confirmed with RETH diagnosis through the evaluation of iodothyronine ratios, crucially the T3/rT3 ratio, invariably low in all patients of the cohort. However, P13, P17, and P18 patients deserve particular comments on their T3/T4 and rT3/T4 ratios (shown in Table 1), clinical presentation, or therapeutic outcome.
P13 is a Turkish man diagnosed at 44 years of age with hypothyroidism due to Hashimoto's thyroiditis (TSH: 100 mU/L, fT4: 0.08 ng/dL, positive anti-TPO, and anti-TG antibodies; Fig. 5). He started treatment with 75 μg LT4/day and 37.5 μg liothyronine (divided into three doses per day). Liothyronine is a sodium-T3 salt, pharmacologically equivalent to T3, that is freely available for prescription in Turkey. After 2 weeks of therapy with an adequate response (TSH lowered to 40 mU/L), he started an LT4 treatment alone (100 μg/day) but he showed an unexpected increase of serum TSH up to 100 mU/L (Fig. 5). Given the suspicion of selective LT4 resistance, combined therapy was re-introduced for 10 weeks, after which TSH drastically decreased (8 mU/L) and fT4 levels normalized. Two further attempts to withdraw liothyronine resulted in corresponding elevations of TSH (Fig. 5). This therapeutic outcome strongly indicated LT4-specific resistance in this patient, along with full sensitivity to T3. The unique T4+T3 combination regime, which this patient received, explains his elevated T3/T4 ratio within the cohort (Table 1). Strikingly, serum fT4 in this patient was, however, low despite the reasonable doses of LT4 received, which is another distinct feature within our cohort. In the absence of intestinal symptoms or any sign of malabsorption, the hormone profile of this patient may suggest LT4-specific impairment of gastrointestinal transport that could be genetically mediated (44). The patient remains with good control under combined T4+T3 therapy.
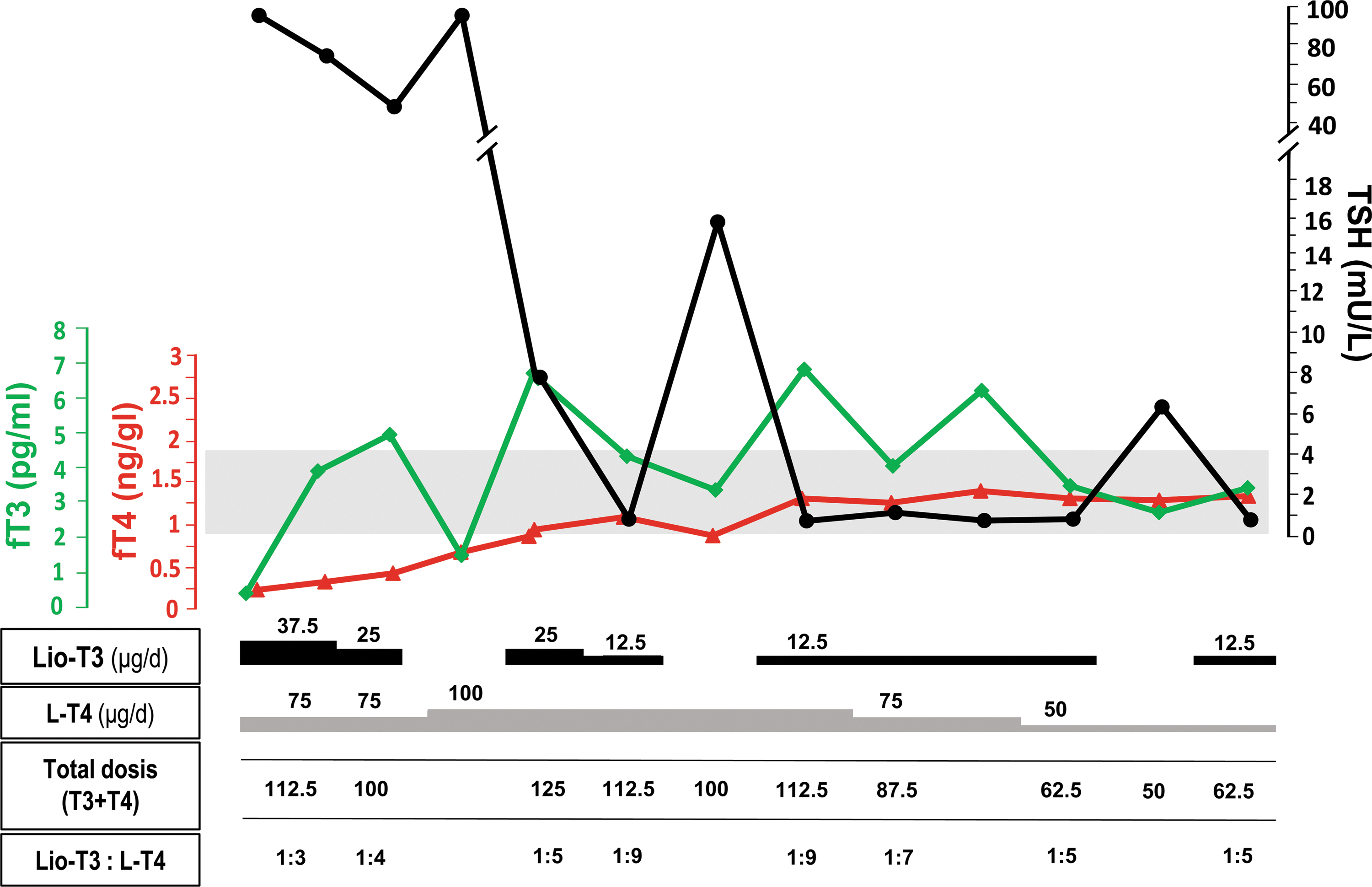
Response to LT4, lio-T3, and combined (T4+T3) treatments of hypothyroidism in patient 13. The figure shows the temporal relationship between fT3 (green line), fT4 (red line), and TSH (black line) under alternating therapeutic regimes with lio-T3, LT4, and T4+T3 treatments. The gray area corresponds to the normal range for fT3, fT4, and TSH, according to the scaled axes. The follow-up course covers the first 3 months from the initial diagnosis and treatment of hypothyroidism due to thyroiditis. Doses of LT4 and lio-T3 are separately indicated, as well as the total iodothyronine (T4+T3) dose and relative ratios. Of note, TSH increased in three separate occasions when lio-T3 was withdrawn to assess sensitivity of the patient to LT4 substitution. TSH could not be normalized with LT4 alone, neither at initial treatment of the severe hypothyroidism nor after normalization and stabilization of fT4 in the patient. fT3, free T3; lio-T3, liothyronine. Color images are available online.
P17 and P18 are the only patients in the series with isolated TSH elevation and a gland in situ of normal size. Further, they are unique among RETH patients based on their T3/T4 ratio. P17 was shown to have hyperthyrotropinemia at 3 years of age. Despite administration of regular doses of LT4 (maximum: 1.8 μg/kg/day), TSH was difficult to normalize because LT4 was not tolerated, showing obvious symptoms of hyperthyroidism. This suggested a central T4 resistance associated with features of peripheral hypersensitivity to T4. He is currently without medication with persistently elevated TSH levels.
Similarly, P18 is a 26 year-old male incidentally diagnosed with permanent TSH elevation. Attempts to normalize TSH through low-dose LT4 (0.7 μg/kg/day) resulted in progressive discomfort with hyperthyroid symptoms (nervousness, insomnia, and diarrhea) for which he was withdrawn from medication after 1 year, even when T4 levels were well within the low-half of the normal range. P18 also had a striking hypersensitivity to T4, even with T4 levels within the normal range, which could represent a subclass of RETH patients showing intolerance to a moderate increase of serum T4. Hypersensitivity to T4, although not strictly defined in humans, is included in the new classification of abnormal sensitivity to thyroid hormone (45). Indeed, peripheral T4 hypersensitivity in association with central (pituitary) insensitivity has been described in rodent models with defects in nuclear T3 co-repressor complexes (46,47). Coincidentally, these 2 patients showed the mildest hormonal derangements at clinical suspicion of RETH, while they also share functional thyroid tissue that is capable of producing T3, perhaps defining a particular etiological subgroup within RETH patients.
Discussion
To our knowledge, this is the first study to identify hypothyroid patients with an overt clinical phenotype and thyroid hormone profiles consistent with RETH. Collectively, 18 patients with hypothyroidism of different etiologies presented with persistently elevated TSH despite iatrogenic hyperthyroidism. After the decrease of LT4 doses, hyperthyrotropinemic euthyroidism could be achieved in all patients. In this phase, the serum profiles revealed iodothyronine ratios (low T3/rT3 and T3/T4, increased rT3/T4) compatible with a defect in T4 to T3 conversion. Interestingly, such a pattern of iodothyronine ratios was independent of serum T4 levels, and was not present in patients with classical RTH-β, supporting their diagnostic and discriminative value.
Defects in iodothyronine deiodinases (D1, D2, and D3) have not been identified as causes of thyroid hormone phenotypes in humans. This contrasts with the overt thyroid hormone derangements shown in rodents with inactive Dio1, Dio2, and Dio3 genes (6,16,17). The only human disorder involving alterations in thyroid hormone deiodination is the rare deficiency of the SECISBP2 (13). Such defects lead to a complex constellation of immune, cutaneous, visual, fertility, and endocrine features (13 –15), which were absent in our patient cohort, suggesting that SECISBP2 was not a candidate susceptibility gene for RETH.
The Dio2(−/−) mouse typically has pituitary resistance to T4, with persistent elevation of serum TSH despite normal T4 levels, indicating a defective feedback in thyrotropes that are unable to adapt TSH synthesis/secretion to serum T4 concentrations (6,7). Primary hypothyroidism induced in these mice through anti-thyroid agents revealed T4-specific resistance, but normal T3-sensitivity to TSH-downregulation, reflecting the local (hypothalamus-pituitary) T4 to T3 conversion deficiency. The thyroid hormone profile of RETH patients strongly suggests a defective D2 deiodination pathway.
In our study, however, patients were wild type for germinal mutations in DIO2. The Ala92-D2 polymorphism was identified in 7 of 18 of RETH patients (40%, as in general population) and was not associated with the hormone profile presented here. This would be expected since, although shown to reduce D2 activity in vitro, the presence of Ala92-D2 in a large cohort of patients who had thyroidectomy, there was a very subtle reduction of serum T3 levels, still within the normal range (18). Nevertheless, Ala92-D2 was recently shown to cause brain hypothyroidism in a mouse model (19), providing a possible explanation for the well-reported, comorbid psychiatric disorders that hypothyroid patients under LT4-alone present when they feel dissatisfied with the treatment (48). Such disease traits were also present in our RETH cohort.
The T4 to T3 conversion defect in RETH may have its origin at different steps of the D2 pathway in the pituitary or the hypothalamus (1). The pituitary adenylate cyclase-activating polypeptide regulates the expression of the Dio2 gene in pituitary thyrotropes, and in hypothalamic tanycytes (49). Polymorphisms in the TRHR gene were associated with serum TSH levels of patients under LT4 because of thyroidectomy (50). The correct synthesis of D2 protein involves the selenocysteine insertion at a position normally coding for the stop codon UGA, which requires four factors: the SECIS element loop at the 3′-UTR of DIO2 (analyzed in our cohort and found wild type), the correct SECISBP2 function, the translation elongation factor eEFSec, and the selenocysteine (Sec) tRNASec, encoded by the TRSP gene (51). The tRNASec needs to interact with several enzymes of the Sec biosynthetic pathway (52), which were recently linked with human pathological conditions (53).
Further, D2 activity is tightly regulated by ubiquitination and deubiquitination, controlling the active half-life of the enzyme in the endoplasmic reticulum (54). D2 ubiquitination disrupts the stability of the D2 dimer, allowing its proteasomal degradation. UBC6 and UBC7 ubiquitin-activating enzymes, and the WSB-1 and TEB-4 ubiquitin ligases were shown to participate in the degradation of D2 at the endoplasmic reticulum (55,56). Ubiquitinated-D2 can be “rescued” from cytosolic proteasomal degradation and reactivated by the USP20 and USP33 deubiquitinases (57). Theoretically, activating or inactivating defects in proteins controlling D2 ubiquitination or deubiquitination, respectively, may be involved in the RETH phenotype, in the absence of DIO2 mutations. Finally, in the intestine, genetic alterations in T4-specific (or preferential) absorption of thyroid hormones, such as the MCT0 transporter, or the new SLC17A4 transporter, may theoretically account for the RETH phenotype in some patients (27,58).
Based on what has been stated earlier, the human RETH may find an explanation in the derangement of one (or more) of the diverse molecular mechanisms and genes involved in the efficiency of the “pituitary D2 pathway,” in charge of adapting the sensitivity of TSH synthesis and secretion to the negative feedback inputs of T4. Epigenetic factors, including non-coding RNAs and histone methylation, might also interfere with the local D3 expression in specific subsets of cells involved in the HPT axis, such as pituitary thyrotropes (59,60).
In conclusion, our work shows a novel clinical phenotype of deranged thyroid hormone metabolism involving T4 to T3 activation and increase of rT3, suggesting a specific defect in D2 action or pathway. The phenotype is dramatically expressed after the patients suffer a primary failure of thyroid hormone synthesis from different etiologies including, but not exclusively, thyroidectomy, and are started on levothyroxine. The RETH patients will benefit from early detection since their reduced capacity for T4 to T3 activation may guide clinicians to avoid rapid and aggressive normalization of TSH in these particular hypothyroid patients. In RETH, a mild hyperthyrotropinemia may be acceptable to avoid iatrogenic hyperthyroidism. Most importantly, the calculation of serum iodothyronine ratios, specifically low T3/rT3 ratio, may represent a good biomarker to guide clinicians toward the implementation of T4+T3 treatment in a targeted and fully comprehensive way in the future. The multiplicity of mechanisms that may underlie this novel disorder of thyroid hormone regulation warrants the implementation of powerful genomic techniques to unravel its possible genetic determinants.
Footnotes
Acknowledgments
The authors thank all patients and/or their parents for their close collaboration in the study, and Mrs. Mercedes Tanarro and Mrs. Elisa Pulido for their excellent technical assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by funding from the Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (grant PI16/00830 to J.C.M.) and by the Rare Diseases Networking Biomedical Research Centre (CIBERER).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1