
Abstract
Background:
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factor is a key regulator of cell survival, proliferation, and gene expression. Although activation of NF-κB signaling in thyroid follicular cells after thyrotropin (TSH) receptor (TSHR) engagement has been reported, the downstream signaling leading to NF-κB activation remains unexplored. Here, we sought to elucidate the mechanisms that regulate NF-κB signaling activation in response to TSH stimulation.
Methods:
Fisher rat-derived thyroid cell lines and primary cultures of NF-κB essential modulator (NEMO)-deficient mice thyrocytes were used as models. Signaling pathways leading to the activation of NF-κB were investigated by using chemical inhibitors and phospho-specific antibodies. Luciferase reporter gene assays and site-directed mutagenesis were used to monitor NF-κB-dependent gene transcriptional activity and the expression of thyroid differentiation markers was assessed by reverse transcription quantitative polymerase chain reaction and Western blot, respectively. Chromatin immunoprecipitation (ChIP) was carried out to investigate NF-κB subunit p65 DNA binding, and small interfering RNA (siRNA)-mediated gene knockdown approaches were used for studying gene function.
Results:
Using thyroid cell lines, we observed that TSH treatment leads to protein kinase C (PKC)-mediated canonical NF-κB p65 subunit nuclear expression. Moreover, TSH stimulation phosphorylated the kinase TAK-1, and its knockdown abolished TSH-induced NF-κB transcriptional activity. TSH induced the transcriptional activity of the NF-κB subunit p65 in a protein kinase A (PKA)-dependent phosphorylation at Ser-276. In addition, p65 phosphorylation at Ser-276 induced acetyl transferase p300 recruitment, leading to its acetylation on Lys-310 and thereby enhancing its transcriptional activity. Evaluation of the role played by NF-κB in thyroid physiology demonstrated that the canonical NF-κB inhibitor BAY 11-7082 reduced TSH-induced expression of thyroid differentiation markers. The involvement of NF-κB signaling in thyroid physiology was confirmed by assessing the TSH-induced gene expression in primary cultures of NEMO-deficient mice thyrocytes. ChIP and the knockdown experiments revealed that p65 is a nuclear effector of TSH actions, inducing the transcripcional expression of thyroid differentiation markers.
Conclusions:
Taken together, our results point to NF-κB being a pivotal mediator in the TSH-induced thyroid follicular cell differentiation, a relevant finding with potential physiological and pathophysiological implications.
Introduction
Thyrotropin (TSH) is the main physiological regulator in thyroid follicular cell proliferation and differentiation, whose action is mediated through G-protein-coupled TSH receptor (TSHR) activation (1). In fact, the TSH-regulated thyroid-differentiated phenotype relies on the expression of several genes implicated in thyroid hormone biosynthesis, with most TSH physiological actions involving the Gαs-activated adenylyl cyclase/cAMP signaling cascade, followed by protein kinase A (PKA) activation (2,3). However, other signaling pathways activated by cAMP are PKA independent, such as those required for TSH-induced thyrocyte proliferation (4,5). Concomitantly with Gαs dissociation after TSHR engagement, Gβγ dimers are released. This leads to phosphatidylinositol 3-kinase (PI3K) activation, which plays a central role in controlling thyroid cell proliferation and differentiation (6). Further, TSH stimulates the Gαq/β-type phospholipase C (PLC-β) cascade and protein kinase C (PKC) activation, promoting thyrocyte cell growth (7). Uncharacterized Gαq-dependent pathways have been reported to be also required for iodide organification and thyroid hormone secretion (8).
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factor is a key regulator of immune response, cell survival, proliferation, and gene expression (9). NF-κB is composed of homo- and heterodimers of five different subunits, including p65, RelB, c-Rel, p50, and p52. The most ubiquitously expressed NF-κB heterodimer is composed of p65 (also known as RelA) and p50 subunits, with p65 being the main transcriptional activator. In unstimulated cells, transcriptionally active NF-κB subunits are sequestered in the cytoplasm as inactive complexes bound to members of the NF-κB inhibitor (inhibitory proteins of κB family [IκB]) family (9). In the canonical pathway, extracellular signals activate the cytoplasmic IκB kinase (IKK) complex, which is composed of IKK-α, IKK-β, and NF-κB essential modulator (NEMO), thereby inducing IKK-β-mediated phosphorylation and proteasomal degradation of IκBs, in particular IκB-α. As a consequence, the released NF-κB dimer translocates to the nucleus, binds to its cognate DNA sequences, commonly referred to as κB sites, in the promoter of target genes, and regulates transcription through the recruitment of coactivators and corepressors (9).
NF-κB has emerged as an important player in thyroid cancer, as activation of the NF-κB signaling pathway in thyroid neoplastic cells has been linked to cell proliferation, invasiveness, angiogenesis, resistance to apoptosis, and the maintenance of transformed phenotypes (10). However, the role of the NF-κB pathway in regulating normal thyroid physiology remains poorly understood. Morshed et al. (11) reported the activation of the NF-κB pathway by stimulating anti-TSHR antibodies in Fisher rat thyroid cell line 5 (FRTL-5) thyroid cells, although the mechanism mediating this activation has not yet been investigated. Further, activation of different Toll-like receptors (TLRs) induced NF-κB signaling in normal thyroid cells (12,13), and we have previously reported that TLR4 activation enhanced the expression of different proteins implicated in thyroid hormone biosynthesis involving NF-κB signaling (14,15). In addition, we observed the presence of autoregulatory S-nitrosylation-mediated mechanisms that may counter-control the transcriptional activity of NF-κB in response to TSHR activation (16). Of note, Reale et al. (17) demonstrated that thyroid-specific NEMO-deficient mice develop a state of hypothyroidism as a result of dramatic thyroid follicular cell loss, in response to the activation of apoptotic mechanisms. In this study, we sought to elucidate the molecular mechanisms that trigger NF-κB signaling activation in response to TSH, as well as investigate the role of NF-κB as a transcriptional mediator of the TSH-regulated thyroid differentiation phenotype.
Materials and Methods
Plasmids
The pTPO-Luc reporter gene containing rat thyroperoxidase (TPO) promoter (−429 to +3 bp), the pNIS-Luc reporter gene containing rat NIS promoter (−2854 to +13 bp), the pTg-Luc reporter gene containing rat thyroglobulin (Tg) promoter (−668 to +36 bp), and the pDuOX2-Luc reporter gene containing human dual oxidase 2 (DuOX2) promoter (−563 to +141 bp) linked to luciferase (Luc) gene were as described elsewhere (18 –21). The NF-κB reporter gene (pNF-κB-Luc), the paired box gene-8 (Pax8) reporter gene (5xPax8-Luc), and the cAMP-responsive gene (5xCRE-Luc) were as previously reported (22 –24). The normalization reporter pCMV-β-galactosidase was obtained from Promega (Madison, WI). Expression vectors encoding wild-type human TSHR, HA-tagged S177A/S181A human IKK-β, S32A/S36A porcine IκB-α, flag-tagged wild-type and S276A human p65, HA-tagged wild-type, and D1485A/I1486L human p300 (p300ΔHAT) were as reported (25 –31).
Cell culture
The Fisher rat-derived thyroid cell lines FRTL-5 and PCCl3 were cultured in Dulbecco's modified Eagle's medium (DMEM)/Ham F-12 medium supplemented with 5% calf bovine serum (Thermo-Fisher Scientific, Waltham, MA), 1 mIU/mL bovine TSH (National Hormone and Peptide Program, Torrance, CA), 10 μg/mL bovine insulin, and 5 μg/mL bovine transferrin (Sigma-Aldrich, Saint Louis, MO) (32). When cells reached 60–70% confluence, they were cultured in the same media for 5 days without TSH but containing 0.2% calf serum, before treatment with 0.5 mIU/mL TSH for different periods. Cells were preincubated with chemical inhibitors for 1 hour before TSH stimulation. The inhibitor concentration was selected on the basis of preliminary dose-response curves, as the highest dose that did not significantly affect cell viability, as determined by the Trypan blue dye exclusion assay.
Genotyped 5-month-old mice (five animals per group) were anesthetized and sacrificed, and the thyroid lobes were dissected as previously described (17). Follicles were seeded and cultured in F12 medium (EuroClone, Milan, Italy) supplemented with 10% Nu-Serum IV (BD BioSciences, San Jose, CA), 10 ng/mL somatostatin (Sigma-Aldrich), and 2 ng/mL glycyl-histidyl-lysine acetate (Sigma-Aldrich). The primary thyrocytes were treated with 1 mIU/mL TSH (Sigma-Aldrich) for 12 hours. Animal procedures were conducted as indicated in the Italian National Guidelines (Nos. 100/2006 and 116/1992) and the European Directives (EEC Council Directive 86/609, 1987), adhering to the Guide for the Care and Use of Laboratory Animals (National Institute of Health, 1996). The animal protocol was approved by the Animal Ethics Committee of the Biogem Consortium (No. 4713).
Transient transfections and gene reporter assays
Thyroid cell lines seeded into six-well plates at 80% confluence were transiently transfected with 2 μg of luciferase reporter constructs/well or with 1 μg of luciferase reporter-promoter constructs plus 2 μg of the expression vector of interest/well by using Lipofectamine 2000 (Thermo-Fisher Scientific) (33). To evaluate promoter activity, cells were split into 24-well plates at 80% confluence the day after transfection. The next day, the growth medium was replaced by basal media, and transfected cells were starved and treated as stated earlier. To assess transfection efficiency, cells were co-transfected with the normalization reporter β-galactosidase. Luciferase activity was measured by using the Luciferase Assay System (Promega) according to the manufacturer's instructions, and normalized relative to that of β-galactosidase.
Site-directed mutagenesis
Site-directed mutagenesis was performed by polymerase chain reaction (PCR), with primers carrying the desired mutation using KOD Hot Start DNA polymerase (EMD Millipore, Temecula, CA), which was followed by template plasmid digestion with DpnI (New England Biolabs, Beverly, MA) (34). All constructs were sequenced to verify specific nucleotide substitutions (Macrogen, Seoul, South Korea).
Iodide uptake
Cells were incubated in modified Hanks' balanced salt solution (10 mM HEPES [pH 7.5], 140 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 0.4 mM MgSO4, 0.5 mM MgCl2, 0.4 mM Na2HPO4, 0.44 mM KH2PO4, and 5.6 mM glucose) containing 20 μM NaI supplemented with 10 mCi/mL carrier-free 125I-iodide (PerkinElmer Sciences, Boston, MA) (35). The radioiodide accumulated in the cells was extracted with 95% ice-cold ethanol and then quantified in a Beckman Gamma 4000 γ-counter (Beckman Coulter, Fullerton, CA). The amount of DNA was determined by the diphenylamine method after trichloroacetic acid precipitation (36).
Immunofluorescence
Cells seeded into 96-well plates were fixed in 2% paraformaldehyde and stained with 2 μg/mL mouse monoclonal anti-p65 antibody (sc-8008; Santa Cruz Biotechnology, Santa Cruz, CA) in phosphate buffered saline containing 0.2% bovine serum albumin and 0.1% Triton X-100 (37). Secondary staining was performed with 4 μg/mL goat polyclonal antimouse Alexa Fluor 594-conjugated antibody (A-11005; Molecular Probes, Eugene, OR), and nuclear DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes). Plates were mounted with FluorSave Reagent (EMD Millipore), and images were acquired on a Nikon Eclipse TS100 inverted microscope (Nikon, Melville, NY). Image quantification was performed by using ImageJ Image Software (National Institutes of Health, Bethesda, MD).
Reverse transcription quantitative polymerase chain reaction
Total RNA purification, complementary DNA (cDNA) synthesis, and quantitative PCR (qPCR) were performed as described (38). The rat gene-specific primer sets were as follows: sodium/iodide symporter (NIS) 5′-GCTGTGGCATTGTCATGTTC (F) and 5′-TGAGGTCTTCCACAGTCACA (R), Tg 5′-GAATTGCTGGCAGATGTTCAG (F) and 5′-GGGCACTGAGCTCCTTGTAG (R), TPO 5′-GGAAGCAGATGAAGGCTCTG (F) and 5′-CGGTGTTGTCACAGATGACC (R), DuOX2 5′-CAAATCGTCCATGGGTGCC (F) and 5′-TCCACAGTTGTCAGAAATAG (R), IκB-α 5′-CCGAGACTTTCGAGGAAATACC (F) and 5′-GAGCGTTGACATCAGCACC (R), and Ribosomal Protein L19 5′-GCGGATTCTCATGGAGCACA (F) and 5′-TCTCCTCCTTCTTGGCTTGGA (R). Mouse gene-specific primer sets were as previously described (17). Relative changes in gene expression were calculated by using the 2−ΔΔCt method normalized against housekeeping genes. All amplification efficiencies ranged between 95% and 110%. The specificity of the reactions was determined by a melting curve analysis. Specific target amplification was confirmed by Sanger's sequencing (Macrogen).
Western blot
Sodium dodecyl sulfate polyacrylamide gel electrophoresis, electrotransference to nitrocellulose membranes, and immunoblotting were carried out as previously described (39). Membranes were blocked and incubated with primary antibodies diluted in 1% skimmed milk, 0.1% Tween 20, and Tris-buffered saline overnight at 4°C. Polyclonal antirat NIS and antihuman DuOX antibodies were as described elsewhere (40,41). Monoclonal anti-TPO (sc-374045), polyclonal anti-TSHR (sc-13936), polyclonal anti-IκB-α (sc-371), and monoclonal anti-Histone H1 (sc-8030) antibodies were from Santa Cruz Biotechnology. Monoclonal anti-p65 (No. 6956), polyclonal anti-phospho p65 (Ser-276; No. 3037), polyclonal anti-acetyl p65 (Lys-310; No. 3045), monoclonal anti-TAK1 (transforming growth factor-β-activated kinase 1; No. 5206), polyclonal anti-phospho TAK1 (Thr-184/187; No. 4531), monoclonal anti-phospho-IκB-α (Ser-32; No. 2859), monoclonal anti-IKK-β (No. 8943), monoclonal anti-IKK-α (No. 11930), and monoclonal anti-phospho-IKK-α/β (Ser-176/180; No. 2697) were from Cell Signaling (Beverly, MA). Polyclonal anti-Tg (A0251), monoclonal anti-α-Tubulin (T6074), and monoclonal anti-E-Cadherin (No. 610182) were obtained from Dako Corporation (Carpinteria, CA), Sigma-Aldrich, and BD Transduction Laboratories (San Jose, CA), respectively. After washing, membranes were further incubated with horseradish peroxidase-labeled secondary antibodies (Santa Cruz Biotechnology) or IRDye 680 RD/800 CW-linked secondary antibodies (LI-COR Biotechnology, Lincoln, NE). The band intensities were quantified densitometrically by using ImageJ Image Software. Equal loading was assessed by stripping and probing the same membrane with the indicated loading controls.
Chromatin immunoprecipitation
Cells were crosslinked in culture media containing 1% formaldehyde, and the nuclei were purified and lysed in 50 mM Tris-HCl (pH 8), 10 mM EDTA, and 1% sodium dodecyl sulfate (42). Genomic DNA was fragmented by sonication and 10-fold diluted in IP Dilution Buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and 0.5% Nonidet P-40. Immunoprecipitation was performed with 2 μg nonspecific mouse IgG or mouse monoclonal anti-p65 antibody (sc-8008; Santa Cruz Biotechnology). Immune complexes were purified with Protein A/G PLUS-Agarose (Santa Cruz Biotechnology), and they were preblocked with sonicated salmon sperm DNA. Immunoprecipitates were washed four times with IP Dilution Buffer supplemented with 0.1% SDS; twice with High Salt IP Wash Buffer containing 50 mM Tris-HCl (pH 7.5), 500 mM NaCl, 5 mM EDTA, 0.1% SDS, and 1% Triton X-100; and once with 10 mM Tris-HCl (pH 8) and 1 mM EDTA. DNA was purified by using Chelex-100 (Bio-Rad, Richmond, CA). Immunoprecipitated DNA was quantified by qPCR, as mentioned earlier. The promoter-specific primer sets were as follows: NIS 5′-AGATTGCAGCTGGCAAGTGC (F) and 5′-AGAAGGTGTTTGGCCTCTTGG (R); Tg 5′-GCTAGCCTCACATTTCTTGTCC (F) and 5′-CTGTCCCCTACTCAAATGATGG (R); TPO 5′-CTGGACTGGATAGAGAAGTGG (F) and 5′-CTCTCTGGAGACTTGGTTACC (R); and DuOX2 5′-GGTAGCAGGGTCCTGTCTGG (F) and 5′-GTTCGGGTTCAACAGTCTTCC (R). The relative fold of increase was calculated according to the equation: 2-[(
Small interfering RNA
FRTL-5 cells were transfected with 50 nM small interfering RNA (siRNA) oligonucleotides/well of a six-well plate by using X-tremeGENE siRNA Transfection Reagent (Roche Applied Sciences, Indianapolis IN). After transfection, cells were TSH starved in media supplemented with 5% calf serum for 72 hours. The siRNA specifically targeting rat NF-κB p65 subunit (sc-61876) and rat TAK1 (No. 6317) were obtained from Santa Cruz Biotechnology and Cell Signaling, respectively. Nonspecific control siRNA (sc-37007) was from Santa Cruz Biotechnology.
Statistical analysis
Results are presented as the mean ± standard error of the mean of at least three independent experiments. Statistical tests were performed by using Prism 3.0 software (GraphPad Software, La Jolla, CA). Multiple-group analysis was conducted by one-way analysis of variance and Newman–Keuls multiple-comparisons post hoc test. Comparisons between two groups were made by using the paired Student's t test. Differences were considered significant at p < 0.05.
Results
TSH activates the canonical NF-κB signaling pathway
Degradation of inhibitory molecules of the IκB family and nuclear accumulation of transcriptionally active NF-κB subunits are hallmarks of NF-κB signaling activation (9). Previously, we found that TSH stimulation of starved FRTL-5 promoted a time-dependent degradation of the IκB-α inhibitor and a concomitant nuclear accumulation of the canonical NF-κB subunit p65 (15,16). Therefore, the NF-κB reporter pNF-κB-Luc was used to study the effect of TSH on NF-κB-dependent gene transcription, with the TSH treatment inducing NF-κB-responsive reporter activity in the thyroid cell lines FRTL-5 and PCCl3 cells (Fig. 1A). Assay specificity was verified by inhibiting TSH-induced pNF-κB-Luc activity in the presence of the IKK-β-specific pharmacological inhibitor BAY 11-7082 (43) (Fig. 1A). Overexpression of the nonphosphorylatable dominant negative mutants S177A/S181A IKK-β (dnIKK-β) or S32A/S36A IκB-α (dnIκB-α) abrogated TSH-induced NF-κB-dependent reporter activity in FRTL-5 cells (Fig. 1B). Moreover, we corroborated the activation of the canonical NF-κB pathway by confirming the phosphorylation of the IKK-α and IKK-β kinases (Ser-176/180) in response to TSH (Fig. 1C). The TSH treatment induced phosphorylation of IκB-α at Ser-32 (Fig. 1D), a prerequisite for ubiquitin-dependent proteasomal degradation, which eventually leads to an increased expression of the p65 subunit in the nucleus of the FRTL-5 cells.
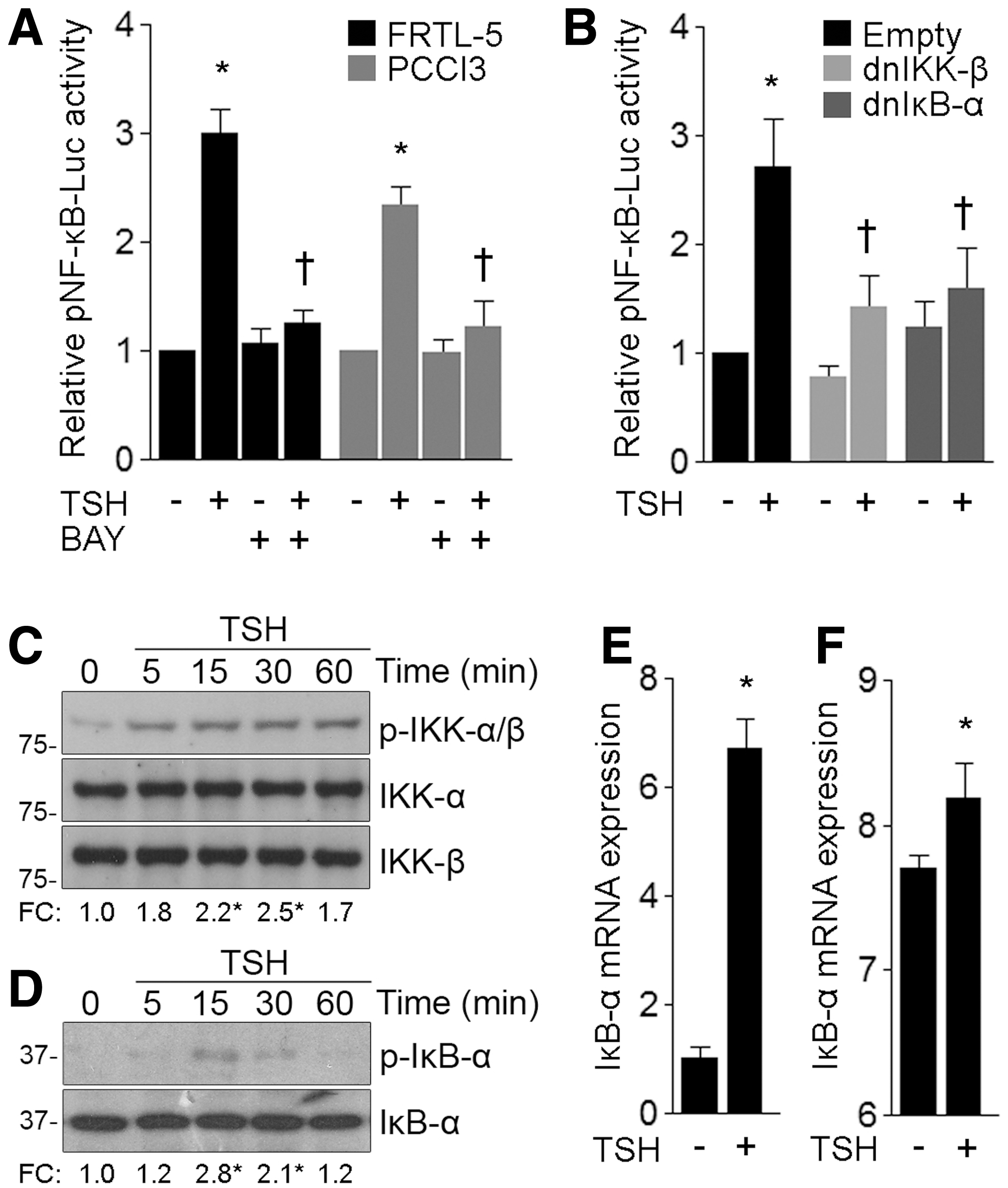
TSH activates NF-κB signaling. (
Unrestricted NF-κB activity relies on the principle that negative signaling regulators are themselves tightly regulated NF-κB targets. In particular, IκB-α is one of the earliest NF-κB-induced target genes and it plays an important role in NF-κB response termination (44). We demonstrated that TSH stimulation increased IκB-α messenger RNA (mRNA) expression in FRTL-5 cells (Fig. 1E). Moreover, differential gene expression data analysis of TSH-treated FRTL-5 cells (45) revealed that IκB-α mRNA was rapidly upregulated after TSH stimulation (Fig. 1F). The biological pathway analysis, which was carried out by using ingenuity pathway analysis (Qiagen Bioinformatics), revealed that the transcriptional program regulated by NF-κB was significantly repressed after long-term TSH stimulation (Supplementary Fig. S1). Taken together, our results indicate that NF-κB-regulated gene expression is promptly activated in response to TSH treatment. Further, NF-κB signaling activation was counter-balanced by the TSH-stimulated negative regulators that terminated signaling.
The PKA and PKC signaling pathways are required for TSH-induced NF-κB-dependent gene expression
Using the NF-κB responsive reporter pNF-κB-Luc, we investigated the TSH-induced signaling pathways involved in the activation of NF-κB-dependent gene transcription, and we observed that the PKA inhibitors H89 and KT5720 inhibited TSH-induced NF-κB-reporter activity (Fig. 2A). In addition, a similar inhibitory effect was observed in the presence of the broad-spectrum PKC inhibitor chelerythrine (CHE) (Fig. 2A). However, no significant effect was found in the presence of the PI3K/AKT pathway inhibitors LY294002 or iAKT1/2. Using Western blot analysis, we demonstrated that TSH-promoted nuclear accumulation of the p65 subunit was significantly repressed after coincubation with the PKC inhibitor CHE, suggesting that a mechanism involving PKC signaling mediates p65 nuclear accumulation in response to TSH signaling (Fig. 2B). The PKA inhibitor H89 did not significantly affect TSH-stimulated p65 nuclear recruitment (Fig. 2B). In line with this, immunofluorescence analysis revealed that the PKC inhibitor CHE, but not the PKA inhibitor H89, substantially reduced TSH-induced p65 nuclear accumulation (Supplementary Fig. S2).

TSH-induced NF-κB activation involved PKA and PKC signaling pathways. (
To further test the cooperative actions of PKA and PKC signaling in TSH-induced NF-κB activation, we assessed the activation of the NF-κB-dependent gene transcription in response to toxic thyroid nodule-identified TSHR gain-of-function mutants that constitutively activate the Gαs/adenylyl cyclase signaling pathway (S505R and D619G TSHR) and the Gαq/phospholipase Cβ pathway (A623I and I486F TSHR) (46). The NF-κB-dependent gene transcription was assessed in TSH-deprived FRTL-5 cells overexpressing the wild-type or the aforementioned TSHR mutants, but only the A623I and I486F TSHR-transfected cells displayed a significant increased NF-κB reporter activity compared with wild-type TSHR-expressing cells in the absence of TSH (Fig. 2C). As a positive control, TSH treatment of empty vector-transfected cells induced NF-κB-dependent gene transcription in response to endogenous TSHR activation (Fig. 2C). Taken together, these data suggest that although only PKC activity participates in the nuclear recruitment of p65 in response to TSH stimulation, TSH-induced NF-κB-dependent gene transcription relies on the cross-talk between PKA and PKC signaling pathways after activation of the TSHR-triggered signal transduction system.
TAK1 links TSH-induced PKC signaling and NF-κB activation
Recent evidence has demonstrated that the ubiquitin-dependent kinase TAK1 plays a critical role in IKK complex activation downstream of PKC in G-protein-coupled receptor signaling pathways (47). Here, we found that TSH stimulation induced a time-dependent TAK1 phosphorylation on critical threonine residues (Thr-184/187) located in the activation loop (Fig. 3A). Moreover, blockage of PKC signaling with the broad-range inhibitors calphostin C and GF109203X blocked TSH-induced TAK1 phosphorylation (Fig. 3B). To further analyze the involvement of TAK1 in TSH-activated NF-κB signaling, we studied NF-κB-driven gene expression in TAK1 knockdown FRTL-5 cells. Western blot analysis revealed a significant knockdown of TAK1 protein levels in TAK1 siRNA-transfected cells, compared with those of scrambled siRNA-transfected cells (Fig. 3C). Interestingly, we demonstrated a significant inhibition of TSH-induced NF-κB activity in TAK1 knockdown cells (Fig. 3D). In agreement, TAK-1 knockdown reduced TSH-promoted IκB-α phosphorylation (Fig. 3E), suggesting that TAK1 mediates TSH-induced canonical NF-κB signaling activation downstream of PKC-dependent signaling events.
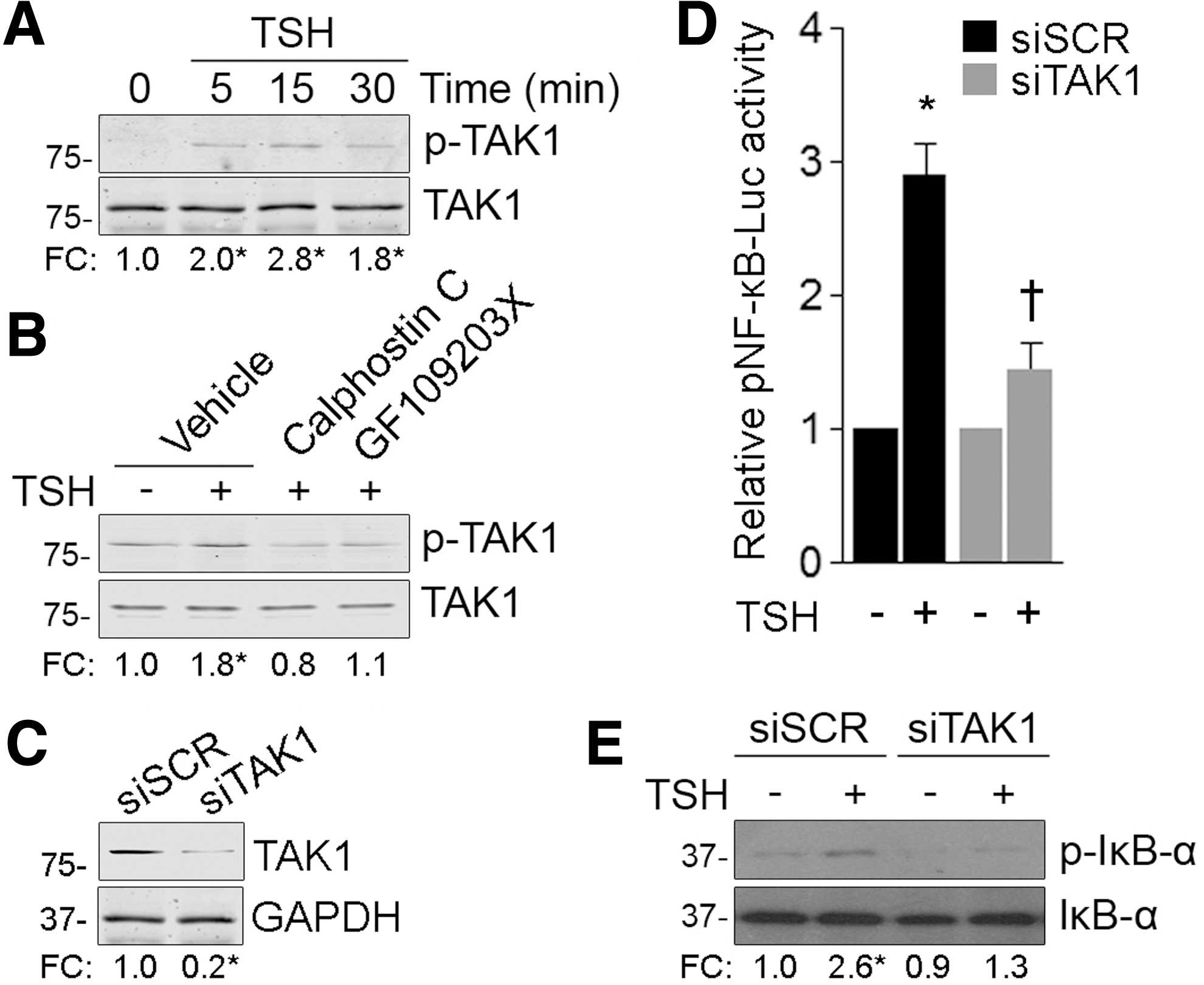
TAK-1 mediates TSH-induced canonical NF-κB signaling downstream PKC-dependent signaling events. (
NF-κB inhibition reduces TSH-stimulated expression of thyroid differentiation markers
To assess the role of NF-κB signaling in TSH-regulated thyroid physiology, we studied the effect of TSH on the expression of thyroid differentiation markers in the presence of the NF-κB inhibitor BAY 11-7082. Iodide transport assays showed that blockage of canonical NF-κB signaling reduced, in a concentration-dependent manner, the TSH-induced iodide accumulation (Fig. 4A). Similar results were observed in the presence of IκB-α phosphorylation and the degradation inhibitor N-α-tosyl-

Canonical NF-κB signaling mediates TSH-stimulated gene expression of thyroid differentiation markers. (
Using a similar experimental approach, we investigated the protein expression of different TSH-regulated thyroid differentiation markers, such as NIS, TPO, DuOX2, and Tg. The Western blot analysis demonstrated that the NF-κB inhibitor BAY 11-7082 reduced TSH-stimulated protein expression of thyroid differentiation markers in a dose-dependent manner (Fig. 4C). We did not find evidence of any significant effect of BAY 11-7082 on TSHR protein expression (Fig. 4D), thus ruling out the possibility that changes in TSHR expression mediated the effect of NF-κB inhibition on TSH-stimulated expression of thyroid differentiation markers. In addition, we examined the activity of the cAMP-responsive vector 5xCRE-Luc under TSH stimulation in the presence of BAY 11-7082. In FRTL-5 cells transiently transfected with 5xCRE-Luc, the NF-κB inhibitor did not significantly affect the activity of the TSH-induced cAMP-responsive vector (Fig. 4E), indicating that NF-κB signaling did not modulate the cAMP-responsive element (CRE)-dependent cAMP-induced transcriptional effect. The specificity of the response of 5xCRE-Luc to TSH stimulation was investigated in the presence of the PKA inhibitor H89 (Fig. 4E).
Consistent with a transcriptional effect, BAY 11-7082-mediated inhibition of NF-κB signaling significantly reduced the TSH-induced mRNA expression of thyroid differentiation markers (Fig. 4F). Next, we further analyzed TSH responsiveness in mouse thyroid primary cultures obtained from thyroid-specific NEMO knockout mice and control littermates, and we observed a significant reduction in the TSH-stimulated NIS, TPO, and Tg mRNA levels in thyrocytes isolated from NEMO-deficient mice compared with control littermates (Fig. 4G). However, no significant differences were observed in TSHR mRNA levels. Importantly, it is known that PKA-dependent TSHR signaling is not compromised in NEMO-deficient thyrocytes, as cAMP-responsive element-binding protein (CREB) phosphorylation in response to TSH stimulation remains unaltered (17). Considering these results together, these data indicate that the TSH-induced gene expression of several proteins involved in thyroid hormonogenesis requires the transcriptional activity of NF-κB, as chemical blockage or the genetic deficiency of canonical NF-κB signaling reduced TSH-stimulated gene expression.
Thyroid-specific gene expression depends on the coordinated action of several transcription factors. In particular, the Pax8 transcriptional activity is essential for thyroid-specific gene expression and thyroid differentiation (48). Therefore, we assessed the involvement of NF-κB signaling in Pax8 expression and transcriptional activity in response to TSH treatment. The blockage of NF-κB signaling did not modulate TSH-upregulated Pax8 protein expression (Fig. 4H) or Pax8 transcriptional activity when assessed by using the Pax8 reporter gene 5xPax8-Luc (Fig. 4I), suggesting a direct transcriptional regulation of NF-κB on the expression of thyroid differentiation markers.
TSH responsiveness relies on cognate κB sites in the promoter of thyroid differentiation markers
To further assess the transcriptional effect mediated by NF-κB in response to TSH stimulation, FRTL-5 cells were transiently transfected with luciferase reporter constructs containing a fragment of the regulatory region of several thyroid differentiation markers. Functional gene promoter analysis revealed that chemical inhibition of canonical NF-κB signaling significantly reduced the TSH-induced transcriptional activity of thyroid differentiation genes (Fig. 5A). In line with this, in silico analysis using the transcription factor binding site prediction software MatInspector (Genomatix Software GmbH) demonstrated the presence of consensus κB sites in the minimal promoter region of Tg (−162 to −150), TPO (−354 to −342), and DuOX2 (−521 to −509); whereas the κB site in the NIS promoter was located within the enhancer region (−2.382 to −2.370) (15) (Fig. 5B). To test functionally the consensus κB sites, we disrupted by site-directed mutagenesis the κB binding site core identified in the aforementioned genes and assessed their response to TSH stimulation. These results demonstrated that the disruption of κB binding sites significantly reduced transcriptional activity in response to TSH stimulation (Fig. 5C), indicating that the κB sites identified are, indeed, functional, and therefore, transcriptionally active NF-κB subunits may participate in the physiological TSH transcriptional responsiveness.
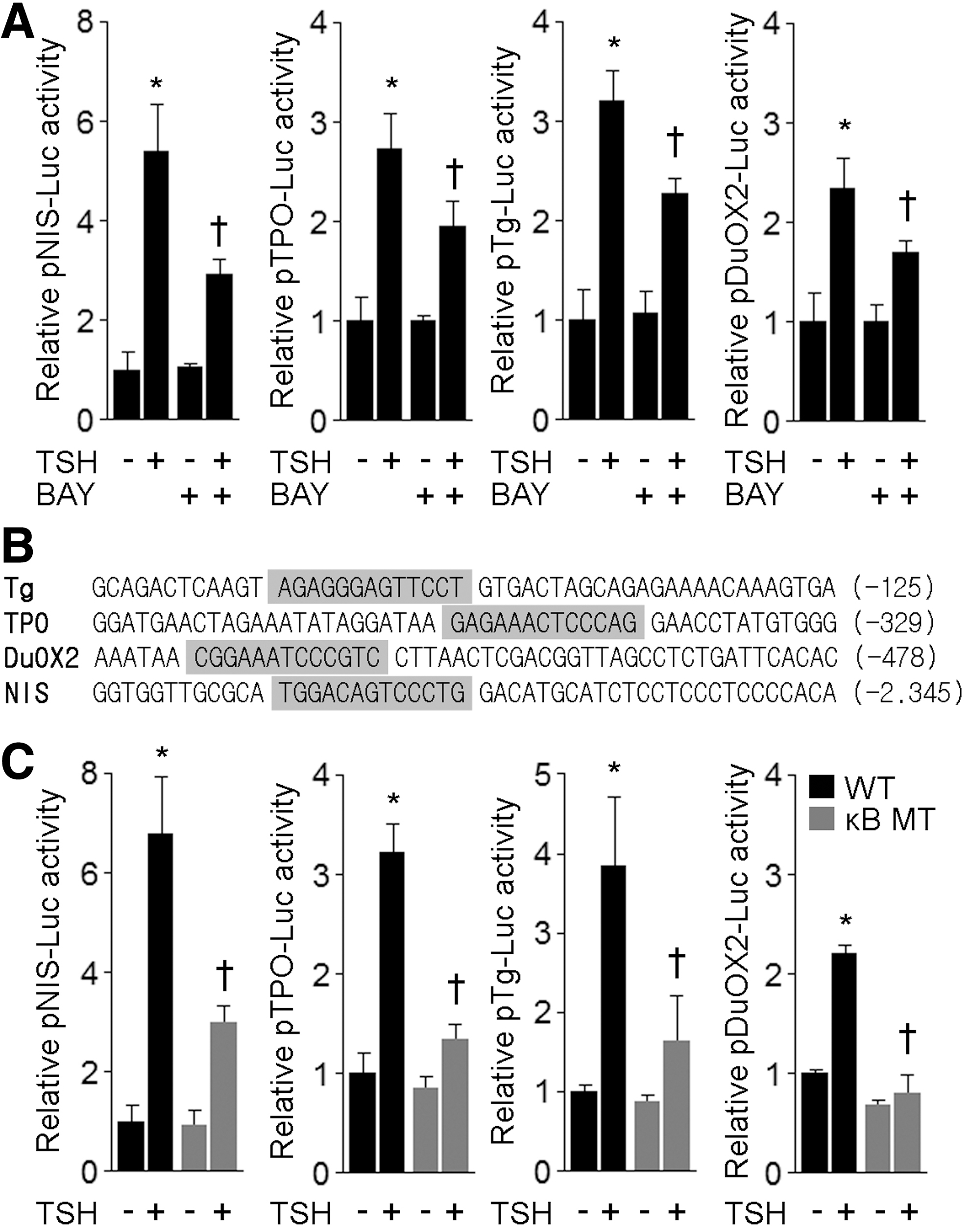
NF-κB binding sites are critical for TSH-stimulated gene expression of thyroid differentiation markers. (
Canonical NF-κB p65 subunit is required for TSH-stimulated gene expression
To determine whether TSH-induced gene expression involves the canonical NF-κB transcriptional effector p65, we used a specific siRNA to knock down endogenous p65 protein expression. Western blot analysis showed that p65 siRNA, but not an unrelated scrambled siRNA, efficiently reduced p65 protein expression (Fig. 6A). However, TSH treatment was ineffective in fully activating NF-κB-dependent gene expression in p65 knockdown FRTL-5 cells (Fig. 6B). Moreover, qPCR analysis revealed that p65 knockdown significantly reduced TSH-induced mRNA expression of thyroid differentiation markers (Fig. 6C). Next, we evaluated the role of p65 as an effector of the TSH-induced transcriptional expression of different genes involved in thyroid hormonogenesis. FRTL-5 cells were cotransfected with scrambled or p65 siRNAs together with the promoter region linked to the indicated luciferase TSH-responsive genes. Under these experimental conditions, a significant reduction of TSH-induced transcriptional activity was demonstrated in p65 knockdown cells (Fig. 6D). These findings indicate that the canonical NF-κB p65 subunit plays a significant role in the TSH-induced gene expression.
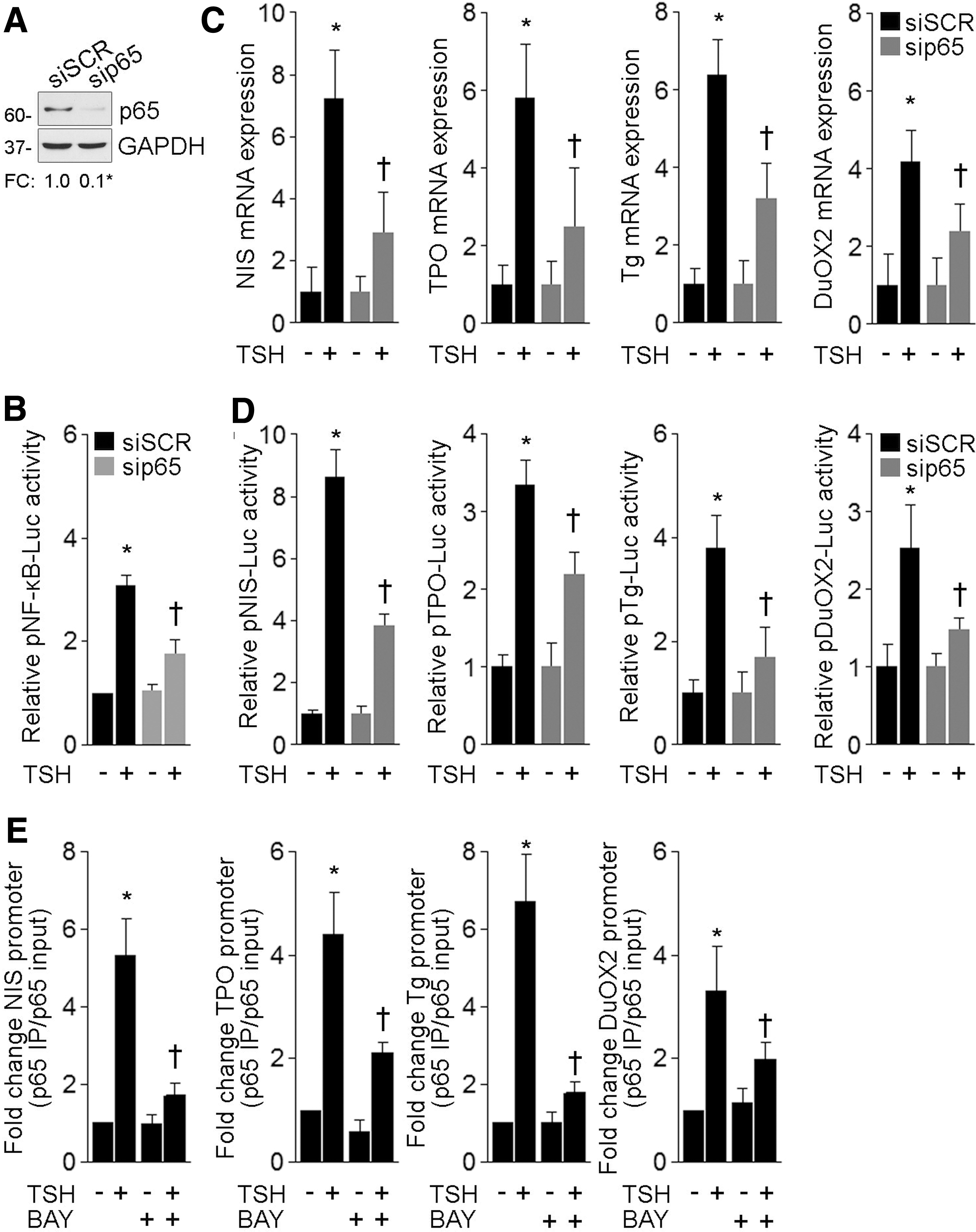
Canonical NF-κB p65 subunit mediates TSH-stimulated gene expression. (
We further examined the association in vivo of p65 to the promoter region of several TSH-regulated thyroid differentiation markers by using chromatin immunoprecipitation (ChIP) assays. Quantitative ChIP analysis revealed that TSH stimulation induced a significant association of p65 to the promoter region of the thyroid differentiation genes Tg, TPO, and DuOX2, and, in turn, we confirmed the interaction of p65 to the NIS enhancer region, as previously reported (15) (Fig. 6E). Interestingly, ChIP analysis showed that enrichment in p65 immunoprecipitates was reduced in the presence of the NF-κB inhibitor BAY 11-7082 (Fig. 6E). Hence, these experiments established that, in response to TSH stimulation, p65 bound to the promoter of different TSH-responsive thyroid differentiation markers (where the functional κB consensus binding sites were identified) to promote gene transcription.
TSH-induced p65 transcriptional activity is stimulated on PKA-mediated phosphorylation at Ser-276
Phosphorylation plays a key role in regulating NF-κB activity. In particular, PKA-mediated p65 phosphorylation at Ser-276 promotes its transcriptional activity (49). Using a phosphospecific antibody, we demonstrated that TSH treatment promoted a significant phosphorylation at Ser-276 (Fig. 7A). Moreover, we corroborated the involvement of PKA in the TSH-induced p65 Ser-276 phosphorylation by using the PKA inhibitor H89. In agreement with our data indicating that TSH-induced NF-κB transcriptional activity is PKA dependent, H89 treatment significantly inhibited TSH-stimulated phosphorylation at Ser-276 (Fig. 7A). These results indicate a potential relationship between p65 phosphorylation and PKA-dependent TSH-induced NF-κB transcriptional activity.
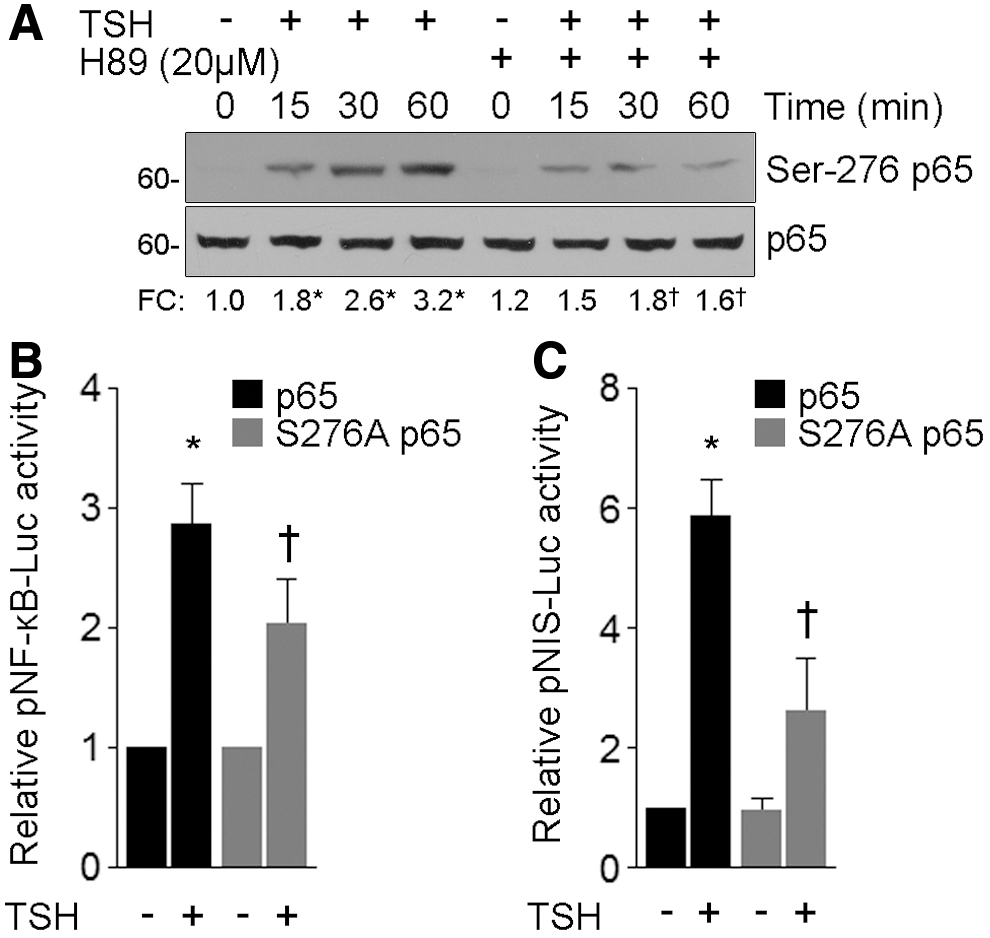
TSH induces PKA-dependent p65 Ser-276 phosphorylation. (
Several reports have demonstrated that PKA-dependent p65 Ser-276 phosphorylation increases p65 affinity for κB enhancer elements and stimulates NF-κB-regulated gene transcription by facilitating the recruitment of the transcriptional coactivators, such as the CREB-binding protein (CBP) paralog E1A-binding protein (p300) (9). Thus, we further evaluated the requirement of p65 phosphorylation in TSH-induced gene expression, with FRTL-5 cells being cotransfected with the NF-κB reporter pNF-κB-Luc together with expression vectors encoding flag-tagged wild-type or S276A p65. Consistent with PKA inhibition, expression of S276A p65 significantly reduced NF-κB reporter activity in response to TSH treatment (Fig. 7B). While attempting to determine the mechanisms responsible for the NF-κB-regulated TSH-induced thyroid-specific gene expression, we focused our attention on NIS expression as a representative thyroid-differentiation gene. We observed a significant reduction in the response of the NIS gene reporter pNIS-Luc to TSH in cells overexpressing S276A p65 (Fig. 7C). Taken together, our results suggest that PKA-mediated p65 Ser-276 phosphorylation is determinant for TSH-induced gene expression.
TSH stimulation induces Lys-310 p65 acetylation
The assembly of NF-κB with different coactivator and corepressor proteins is crucial in orchestrating gene expression. The transactivation potential of p65 is also regulated by CBP/p300-induced acetylation, and, in particular, p300-mediated Lys-310 acetylation is associated with full NF-κB transcriptional activity (9). Using an antibody targeting p65 acetylated at Lys-310, we demonstrated acetylation of p65 at Lys-310 in response to TSH treatment by Western blot analysis of immunoprecipitated p65 from the nuclear fraction of FRTL-5 cells (Fig. 8A). Thereafter, we evaluated the involvement of the acetyltransferase p300 in the TSH-induced p65-dependent NF-κB-responsive reporter activity. FRTL-5 cells were cotransfected with the reporter pNF-κB-Luc together with expression vectors encoding HA-tagged wild-type (HA-p300) or the acetyltransferase-deficient mutant D1485A/I1486L p300 (HA-p300ΔHAT). HA-p300ΔHAT overexpression significantly repressed TSH-stimulated pNF-κB-Luc reporter activity (Fig. 8B). Consequently, we assessed the role of p65 Lys-310 acetylation in TSH-induced NIS gene expression. Cells were cotransfected with the NIS gene reporter pNIS-Luc along with flag-tagged wild-type p65 or acetylation-deficient p65 mutants, where Lys-310 (K310R p65) or Lys-314 and 315 (K314R/K315R) was replaced by the nonacetylatable residue Arg. We observed a significant reduction in the TSH-stimulated pNIS-Luc reporter activity in the presence of K310R p65 (Fig. 8C). However, no significant difference in the response to TSH was found in cells overexpressing K314R/K315R p65 (Fig. 8C). Considering these results together, our data suggest that the acetyltransferase activity of p300 is required in the TSH-induced NF-κB-mediated expression of thyroid differentiation genes, involving a mechanism dependent on p65 Lys-310 acetylation.
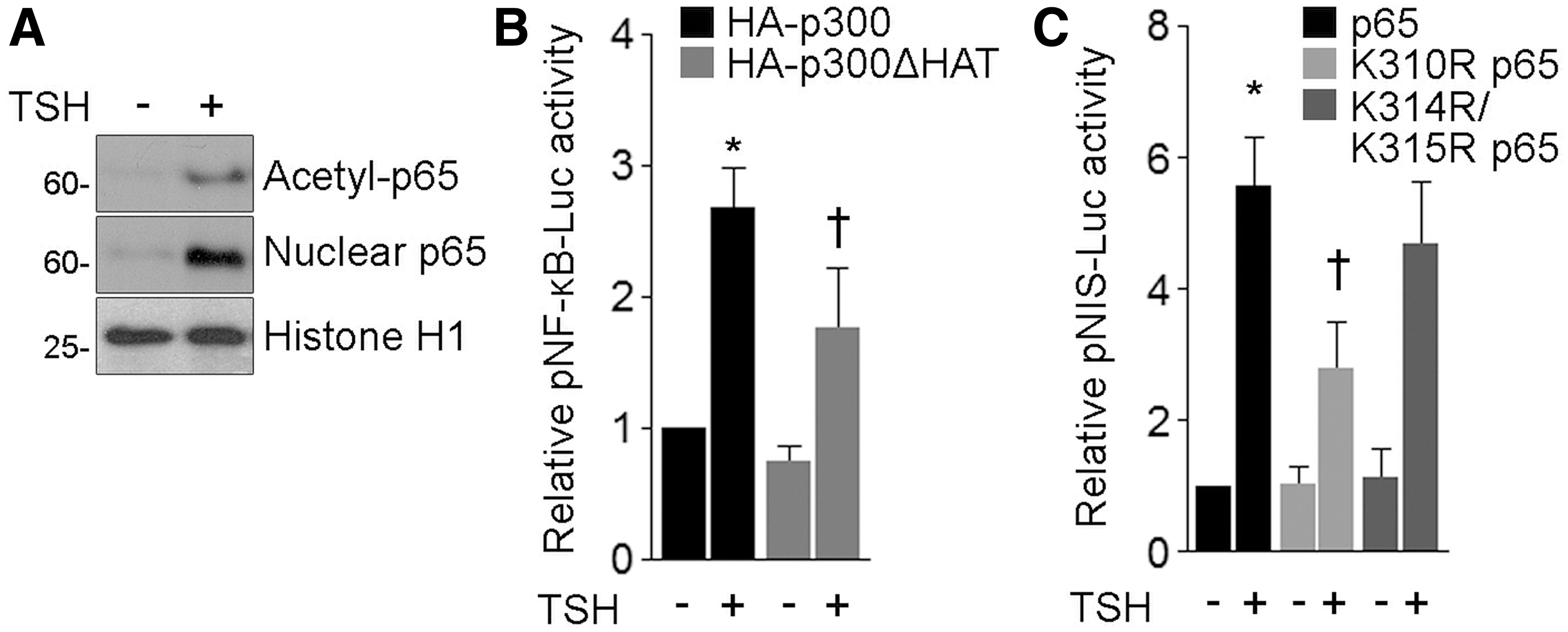
TSH induces p65 Lys-310 acetylation. (
Discussion
Although many investigations have focused on elucidating the TSH-stimulated signaling pathways involved in thyroid cell proliferation and differentiation, little is known about the signaling cascades that regulate thyroid cell differentiation, other than the cAMP/PKA pathway, which is understood to be the main TSH-activated signaling (2,3). The best-characterized PKA substrate is, in fact, the ubiquitous transcription factor CREB, which interacts with CRE sequences in target promoters. Nevertheless, several cAMP-responsive thyroid-differentiation genes do not contain any canonical CRE sequences in their promoter regions (50). Current knowledge suggests that cAMP-dependent gene expression might occur indirectly through regulation of the transcriptional activity of other thyroid transcription factors, such as Pax8 (51,52). Indeed, Sastre-Perona and Santisteban (53) reported a PKA-dependent TSH-induced mechanism that stimulates β-catenin transcriptional activity to regulate Pax8-dependent gene transcription in differentiated thyroid cells. Although the identification of CREB-independent mechanisms still remains to be fully explored, in this study, we provide novel evidence supporting the activation of the canonical NF-κB signaling pathway downstream to TSHR activation in differentiated thyroid cells (Fig. 9).

Schematic representation for NF-κB-dependent TSH-stimulated expression of thyroid differentiation markers. TSH-stimulated PKC signaling triggers TAK-1-dependent IKK-β-mediated phosphorylation and subsequent proteasomal degradation of the NF-κB inhibitor IκB-α. The canonical NF-κB p65 subunit is then translocated to the nucleus and recruited to the promoter of thyroid differentiation markers. Concomitantly, TSH-activated PKA mediates p65 Ser-276 phosphorylation, promoting its interaction with p300, which, in turn, acetylates p65 at Lys-310 and thereby increases p65 transcriptional activity, enhancing the transcription of thyroid differentiation markers. Ac, acetylation; AC, adenylate cyclase; P, phosphorylation.
Several studies have demonstrated the involvement of NF-κB in thyroid physiology and pathology, including autoimmunity and cancer (10). Despite the fact that little is known regarding the role of NF-κB in thyroid cell physiology, several reports have indicated functional NF-κB signaling in normal thyroid cells. Moreover, investigations on the major histocompatibility complex (MHC) class I gene in FRTL-5 cells have shown that its expression is hormonally controlled through the regulation of NF-κB binding to the MHC class I promoter (54,55). It is significant that tumor necrosis factor (TNF)-α-dependent p65-modulated target gene expression relies on the presence of TSH-stimulated PKA activity (56,57). Morshed et al. (11) reported the activation of the NF-κB signaling pathway measured as p65 Ser-536 phosphorylation in response to TSH or anti-TSHR-stimulating antibody treatment in FRTL-5 cells. Also, evaluation of the transcriptional mechanism triggered by the bacterial endotoxin lipopolysaccharide led us to identify the role of the canonical NF-κB p65 subunit in the transcriptional regulation of NIS and TPO gene expression (14,15). More recently, Reale et al. (17) highlighted the role of canonical NF-κB signaling in thyroid physiology. Using a thyroid-specific NEMO-deficient mouse model, these authors revealed that canonical NF-κB signaling is required for thyroid follicular cell survival and the maintenance of thyroid differentiation (17). Other studies have demonstrated functional TSHR expression in extrathyroid tissues, such as liver, adipose tissue, and bone. Experimental evidence has supported TSH-mediated gene expression in several nonthyroid tissues involving the transcription factor NF-κB. Specifically, in adipose tissue, TSH action has been reported to regulate adipogenesis and lipolysis during development (58,59). Further, Antunes et al. (60) observed that TSH-induced interleukin-6 expression involves NF-κB activation in human adipocytes. Importantly, TSHR expression occurs in precursors and mature osteoclasts, and TSH stimulation negatively regulates bone resorption by inhibiting osteoclast differentiation and survival through the modulation of NF-κB signaling (61).
In canonical NF-κB signaling, activation of the IKK complex is essential for inducing IκB-α degradation and subsequent nuclear translocation of NF-κB complexes, in particular, the p65/p50 dimer (9). Therefore, deciphering cell-type-dependent and signal-specific IKK complex regulation is central to understanding the complexity of NF-κB activation. Significantly, the regulatory subunit NEMO is required for the activation of the canonical NF-κB signaling pathway, as NEMO-deficient cells do not activate canonical NF-κB pathway in response to many stimuli. Here, we demonstrated that TSH-induced canonical IKK-β-dependent NF-κB-regulated gene expression involves a synergic crosstalk between the PKA and PKC signaling pathways (Fig. 9), with TSH-activated TAK-1 being a downstream target of PKC signaling and participating in the phosphorylation of the IKK complex and subsequent activation of the canonical NF-κB pathway (Fig. 9). Moreover, TAK1 is also a ubiquitin-dependent mitogen-activated protein kinase kinase (MAPKK) that is involved in the c-Jun N-terminal kinase (JNK) pathway. Interestingly, Hara et al. (62) reported that TSH-induced JNK activation relies on a PKC-dependent pathway involving calcium-independent novel PKC subunits. Thus, TAK1 appears to represent a relevant yet poorly studied kinase in the TSH/TSHR signal transduction pathway in thyroid follicular cells, with TAK1 inhibition promoting apoptosis of B-CPAP papillary thyroid cancer cells by inhibiting the NF-κB signaling pathway (63). Supporting our results, Gagnon et al. (64) reported that PKC-δ inhibition reduced TSH-stimulated IKK-β phosphorylation in human differentiated adipocytes. Moreover, Raychaudhuri et al. (65) revealed that TSH-induced interleukin-6 expression in CD34+ thyroid-associated ophthalmopathy orbital fibroblasts partially relies on phosphoinositide-dependent kinase-1 (PDK1)-dependent PKCβII-activated canonical NF-κB signaling.
Since the canonical NF-κB signaling pathway has been shown to be critical for thyroid follicular cell survival and differentiation (17), here we investigated the role played by the canonical NF-κB transcription factors as physiological mediators of the TSH-induced gene expression of thyroid differentiation markers. Chemical and genetic canonical NF-κB signaling pathway inhibition revealed a pivotal role in TSH-induced thyroid follicular cell differentiation. Moreover, bioinformatic analysis of the promoter region of the different TSH-responsive genes involved in thyroid hormone biosynthesis allowed us to identify the putative consensus NF-κB binding sites, and using site-directed mutagenesis, we demonstrated the involvement of NF-κB inTSH-stimulated gene expression. We focused our investigation on the role of the canonical NF-κB subunit p65 as a TSH-stimulated transcription factor involved in thyroid differentiation, with the ChIP analysis demonstrating the interaction of p65 with the promoter region of thyroid differentiation markers in response to TSH stimulation. Also, siRNA-mediated p65 knockdown reduced the induction of the thyroid differentiation markers in response to TSH treatment. These data, therefore, support the involvement of the transcription factor p65 in the TSH-regulated thyroid differentiated phenotype (Fig. 9). In agreement, with the importance of the canonical NF-κB signaling pathway in thyrocyte differentiation, Reale et al. (66) reported that the environmental thyroid disrupting chemical 2,3,7,8-tetrachlorodibenzo-p-dioxin represses thyrocyte differentiation through an NF-κB pathway-dependent mechanism.
Phosphorylation plays an important role in regulating NF-κB transcriptional activity, mainly by modulating the recruitment of transcriptional coactivators required for gene regulation (9). On assessing the phosphorylation status of p65 in response to TSH treatment, we observed a PKA-dependent p65 Ser-276 phosphorylation, thus establishing a link between p65 phosphorylation and TSH-induced NF-κB transcriptional activity, as the blockage of p65 Ser-276 phosphorylation impaired TSH-stimulated NF-κB-dependent gene expression (Fig. 9). Moreover, considering that p65 Ser-276 phosphorylation promotes its interaction with p300, which, in turn, acetylates p65 at Lys-310 and thereby increases p65 transcriptional activity (49), we demonstrated that TSH induces p65 Lys-310 acetylation (Fig. 9). Importantly, NF-κB acetylation has been reported to regulate the transcriptional potential of NF-κB, the duration of the NF-κB response, and the NF-κB interactions with several transcription cofactors (67). Our results suggest that there are additional but yet to be identified TSH-induced mediators that regulate NF-κB activation, as well as p300 acetyltransferase activity, to modulate site-specific p65 acetylation. Similarly, the transcriptional coactivator p300 has been shown to be crucial for Pax8-induced TPO gene expression (68). The recruitment of p300 by Pax8 may induce localized chromatin remodeling, resulting in the enhancement of Pax8-mediated gene transcription. Also, the inability of the congenital hypothyroidism-causing S48F Pax8 mutant to recruit p300 may explain the loss of its transactivation potential (69).
The evidence presented lays the groundwork for considering the canonical NF-κB signaling pathway to be a relevant player in the complex TSH-stimulated transcriptional network that regulates thyroid follicular cell differentiation. Although we have focused our attention on the participation of the p65 subunit, our results do not exclude the participation of other, less characterized, transcriptionally active NF-κB family members from being downstream mediators of TSH actions. However, we consider that further studies are necessary to address key issues, such as the role of Gαq-dependent PKC isoenzymes in the activation of NF-κB signaling, to obtain a more complete characterization of the NF-κB negative feedback regulatory mechanisms and to identify the molecular mechanisms by which NF-κB activates transcriptional expression of the thyroid differentiation markers. Recently Faria et al. (70) demonstrated that TNF-α-repressed TSH-stimulated NIS gene expression in normal thyroid cells involves canonical NF-κB signaling activation. Thus, NF-κB-regulated gene expression may lead to antagonist effects that rely on context-dependent signaling crosstalk triggering its activation and post-translational regulation. Indeed, we demonstrated that p65 phosphorylation at Ser-536 constitutes a critical step in p65-dependent TPO gene expression in response to lipopolysaccharides (14).
The inappropriate regulation of NF-κB activation has been directly involved in a wide range of human disorders, including cancer, autoimmunity, and neurodegenerative and inflammatory diseases. For example, transcriptomic data analysis has revealed that NF-κB signaling may act as a major regulator of different signaling pathways enriched in the thyroid gland of patients with Graves' disease (71). NF-κB constitutes the central effector downstream in thyroid-specific TNF receptor family member CD40 activation (72). Moreover, NF-κB has been postulated as being a key link in chronic inflammation-associated cancer progression (73). The sustained activation of canonical NF-κB signaling has been identified in differentiated and undifferentiated thyroid carcinomas (74), with NF-κB signaling activation being triggered by different oncogenes involved in thyroid carcinogenesis. Mechanistically, BRAFV600E activates NF-κB signaling independently of the MAPK/ERK downstream pathway (75). Further, we have previously shown that BRAFV600E promotes functional TLR4 overexpression, suggesting that host-derived TLR4 ligands might trigger NF-κB signaling in thyroid carcinomas (76). Significantly, the polymorphism rs2233406 in the NFKBIA gene, which encodes the NF-κB signaling inhibitor IκB-α, has been associated with a tumor progression-prone proinflammatory microenvironment (77). Increased NF-κB activity is associated with thyroid carcinogenesis and tumor progression. In particular, BRAFV600E -activated NF-κB signaling increases tissue inhibitor of metalloproteinases (TIMP-1) expression, which, in turn, triggers PI3K/Akt signaling pathways, increasing cellular proliferation and invasiveness, the inhibition of apoptosis, and resistance to doxorubicin (75,78). In addition, the inhibition of NF-κB signaling potentiates the inhibitory effects of MEK inhibitors, thereby providing an attractive therapeutic strategy for overcoming the resistance to MEK inhibitors for advance thyroid carcinomas (79,80). Therefore, a more complete understanding of the mechanisms regulating NF-κB signaling pathway activation in thyroid pathologies may be of important therapeutic interest, in terms of identifying new therapeutic alternatives.
Footnotes
Acknowledgments
The authors thank Dr. Nancy Carrasco (Vanderbilt University School of Medicine, United States), Dr. Pilar Santisteban (Instituto de Investigaciones Biomédicas Alberto Sols, Spain), Dr. Roberto Di Lauro (Università degli Studi di Napoli Federico II, Italy), Dr. Toshimasa Onaya (University of Yamanashi, Japan), Dr. Franҫoise Miot (Université Libre de Bruxelles, Belgium), Dr. James M. Samet (U.S. Environmental Protection Agency, United States), Dr. Dietmar Spengler (Max Planck Institute of Psychiatry, Germany), Dr. Anna Bigas (Institut Hospital del Mar d'Investigacions Mèdiques, Spain), Dr. Basil Rapoport (University of California Los Angeles, United States), Dr. Michael Karin (University of California San Diego, United States), Dr. Andreas Hecht (Max-Planck-Institute of Immunobiology, Germany), and Dr. Rainer De Martin (Medical University of Vienna, Austria) for providing reagents. They also thank Dr. Pilar Crespo from the Centro de Micro y Nanoscopía de Córdoba (CEMINCO-CONICET)—Universidad Nacional de Córdoba for technical assistance concerning imaging, and also the members of their laboratory for their helpful methodological assistance and critical insights.
Author Disclosure Statement
All the authors declare that no competing financial interests exist.
Funding Information
This work was supported by fellowships and research grants from Consejo Nacional de Investigaciones Científicas y Técnicas, Fondo para la Investigación Científica y Tecnológica—Agencia Nacional de Promoción Científica y Tecnológica (PICT-2008-0890 and PICT-2014-2564 to A.M.M.-R., and PICT-2014-0726, PICT-2015-3839, and PICT-2015-3705 to J.P.N.), Secretaría de Ciencia y Tecnología—Universidad Nacional de Córdoba (30820150100222CB and 33620180100772CB to J.P.N.), Thyroid Cancer Survivors' Association (2015-033 to J.P.N.), Instituto Nacional del Cáncer—Ministerio de Salud y Desarrollo Social, and Latin American Thyroid Society (to J.P.N.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2