
Abstract
Background:
Iodothyronine deiodinase-1 (D1) selenoenzyme regulates the systemic supply of active thyroid hormone (TH). Transient decrease in D1 enzymatic activity is clinically relevant and adaptive in nonthyroidal illness such as fasting or acute illness. However, DIO1 gene defects have not been reported in humans.
Methods:
Genetic analysis was performed using whole-exome sequencing in members of two unrelated families presenting with abnormal serum thyroid function tests. Plasmid constructs containing the two pathogenic DIO1 variants were used for in vitro studies assessing the kinetics of their enzymatic activity. Thyroid function tests were measured in Dio1 heterozygous-null mice.
Results:
We report the novel identification and characterization of two missense DIO1 pathogenic variants (resulting in
Conclusions:
We report the identification and characterization of two missense DIO1 pathogenic variants identified in families with abnormal TH metabolism. This is the first demonstration of inherited D1 deficiency in humans.
Introduction
The Three Iodothyronine Deiodinases (D) are selenoenzymes that modulate thyroid hormone (TH) action by regulating the availability of the active TH, triiodothyronine (T3) (1). T3 is generated via outer ring 5′ deiodination by D1 and D2 of the prohormone thyroxine (T4) and is degraded via inner ring 5 deiodination by D3. Specifically, D1 enzymatic activity modulates the serum levels of T3, while D2 and D3 fine-tune the intracellular availability of T3. D1 is mainly expressed in the liver, kidney, and thyroid gland and has a relatively high Km for its substrates T4 and reverse T3 (rT3). Although systematically sought after, genetic defects in the human deiodinases genes (DIO) have remained elusive.
Materials and Methods
Genetic analysis
The study was approved by the institutional review boards of The University of Chicago, and written informed consents were obtained. No new genomic data were generated and GRCh37/hg19 was used as reference.
To evaluate the genetic diagnosis of the patients, DNA samples from an affected individual from each family were submitted to whole-exome sequencing (Novogene Corp) using SureSelect Human All Exons Kit (Agilent Technology, Santa Clara, CA) and sequenced on a HiSeq2500 system (Illumina, San Diego, CA). Briefly, the resulting paired-end reads (FASTQ files) were aligned to human reference genome GRCh37/hg19 using Burrows–Wheeler alignment tool (2). Variant calling was simultaneously performed with GATK in all the resulting BAM files. Finally, the resulting variants were annotated with ANNOVAR (3). We focused our analysis on rare protein-altering variants (nonsynonymous, InDels, nonsense, and splicing site). Variant frequency was analyzed based on different ethnic subgroups available at the Exome Aggregation Consortium, the NHLBI Exome Sequencing Project Exome Variant Server, and the International Genome Sample Resource 1000 Genomes Project. Single-nucleotide variants were run through nine independent protein pathogenicity predictors: Polyphen-2, SIFT, Mutation Taster, Mutation Assessor, FATHMM, and LRT. Annotation also included information about base conservation and functional predictions from the Combined Annotation Dependent Depletion database and Genomic Evolutionary Rate Profiling score (Table 1). The mean coverage depth of the exome targeted regions was 100x. The respective DIO1 pathogenic variants were confirmed by Sanger sequencing in all subjects of each family. Using the American College of Medical Genetics and Genomics (ACMG) guidelines (4), these variants were curated as pathogenic (N94K: PS3+PM1+PM2+PP1+PP2+PP3+PP4; M201I: PS3+PM1+PM2+PP2+PP3+PP4) (Table 2).
Pathogenic Variants Identified in Two Unrelated Families Presenting with Abnormal Thyroid Hormone Metabolism
Clinical interpretation of genetic variants by ACMG/AMP 2015 guideline (4). PS3: Well-established in vitro or in vivo functional studies supportive of a damage effect on the gene or gene product. PM1: Located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation. PM2: Absent from controls (or at extremely low frequency if recessive) in public database, such as the Exome Sequencing Project, 1000 Genomes Project, Exome or Genome Aggregation Consortium. PP1: Co-segregation with disease in multiple affected family members in a gene definitely known to cause the disease. PP2: Missense variant in a gene that has a low rate of benign missense variation and in which missense variant is common mechanism of disease. PP3: Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.). PP4: Patient's phenotype or family history is highly specific for a disease with a single genetic etiology.
ACMG, American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; gnomAD, Genome Aggregation Consortium; MAF, minor allele frequency.
In Silico Prediction of DIO1 Pathogenic Variants
Based on ANNOVAR guideline.
D: Probably damaging (≥0.909), P: Possibly damaging (≥0.447 and ≤0.909).
GERP and CADD: higher scores are more deleterious.
CADD, Combined Annotation Dependent Depletion database; GERP, Genomic Evolutionary Rate Profiling score.
Plasmid constructs
NH2-terminal FLAG-tagged wild-type (WT) human D1 plasmids (Curcio-Morelli-Bianco Endocrinology 2003) were subcloned in pcDNA3 vector. The pcDNA3-D1-WT construct contains 1861 bp of DIO1 gene and was used as control. The empty vector of pcDNA3 was also used as negative control. The c.282C>A and c.603G>A missense DIO1 pathogenic variants were individually generated using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent) as specified by the manufacturer. The mutagenic primers were designed using the web-based QuikChange Primer Design Program. The site-directed mutagenesis was confirmed by automatic sequencing using the Sanger method (data not shown).
D1 enzymatic activity assay
HEK 293 cells were grown and maintained in DMEM supplemented with 10% fetal bovine serum and used at 70–80% confluence. Using 10 cm plates, the cells were transiently transfected with D1 plasmids (10 μg) and Lipofectamine®2000 (Life Technologies, Carlsbad, CA). A pGFP-expressing plasmid was used to control for transfection efficiency (2 μg per well); GFP was quantified using Quantification Kit (ab235672; Abcam, United Kingdom). At 48 hours after transfection, cells were washed and harvested with sterile PBS and pellets obtained after cold centrifugation at 81.6 g for five minutes; sonication was in 0.25 M sucrose in PE buffer (0.1 M potassium phosphate and 1 mM EDTA) with 10 mM DTT. Sonicated HEK-293 cells transfected with 10 μg empty vector (pCDNA3) were used to determine the background D1 activity. For the D1 activity assay, 125I-T4 (1080–1320 μCi/μg; PerkinElmer) was incubated with 100 μg protein, 1 μM of unlabeled T4, 10 mM DTT, 75,000 cpm 125I-T4, and PE buffer up to 300 μL. Experiments were performed in duplicate or triplicate for each condition. The reaction was incubated at 37°C in water bath for three hours and terminated by addition of 200 μL of horse serum and 100 μL of 50% trichloroacetic acid, followed by centrifugation at 21,000 g for three minutes. After precipitation, 360 μL of the supernatant was removed and quantified by automatic gamma counter (Wizard2; PerkinElmer). Results are shown as femtomoles of T3 per minute per milligram protein (fmol/min/mg protein). To evaluate D1 velocity in cells expressing WT or mutant pD1, various amount of unlabeled T4 concentrations (0.1, 0.2, 0.5, 1, 2, 5, and 10 μM), together with 75,000 cpm 125I-T4, were also applied in the study. All conditions were performed in three independent experiments.
Experimental animals
Procedures carried out on mice were approved by the University of Chicago Institutional Animal Care and Use Committee. Dio1-KO mice were generated and housed as previously described (5,6). Experiments were performed on P60–80 adult male WT, Dio1-Het, and Dio1-KO mice whose genotypes were confirmed as previously described (5). Blood samples were collected; serum was separated and frozen before measurement of total rT3 and T3 concentrations as described (7). Seven to 10 animals were used in each group.
Statistics
Statistical analysis used one-way analysis of variance for multiple comparisons using GraphPad Prism Software (GraphPad, Inc., San Diego, CA). Results are represented as mean ± SE. At least, three independent experiments were performed for each condition. p Value ≥0.05 was not considered significant.
Results
Case presentations
We report two families (Fig. 1A) referred for investigation of unexplained and discrepant thyroid function tests. In family 1, the proband (III-3), a three-year-old girl with Down syndrome, born from nonconsanguineous parents of Hispanic origin, was found to have slightly elevated serum thyrotropin (TSH) and positive thyroperoxidase antibodies, commonly associated with Down syndrome (8). However, her sisters, mother, maternal aunts, and grandmother exhibited elevated serum rT3 levels and rT3/T3 ratios when compared with unaffected family individuals (Fig. 1B). The whole-exome sequencing revealed that all family members with these characteristic thyroid tests harbored a rare heterozygous pathogenic variant in DIO1 gene (NM_000792_c.282C>A rs754694815, p.Asn94Lys, herein referred as N94K). In the second family (Fig. 1A), a 15-year-old girl (II-2), born from nonconsanguineous Jewish parents, was referred for resistance to TSH; however, no mutations were identified in the TSH receptor gene. It was noted that the proposita and her mother had serum rT3 levels and rT3/T3 ratios higher than other family members (Fig. 1B). A second rare heterozygous pathogenic variant in DIO1 (NM_000792_c.603G>A rs201420507, p.Met201Ile, herein referred as M201I) was identified in the proposita and her mother.
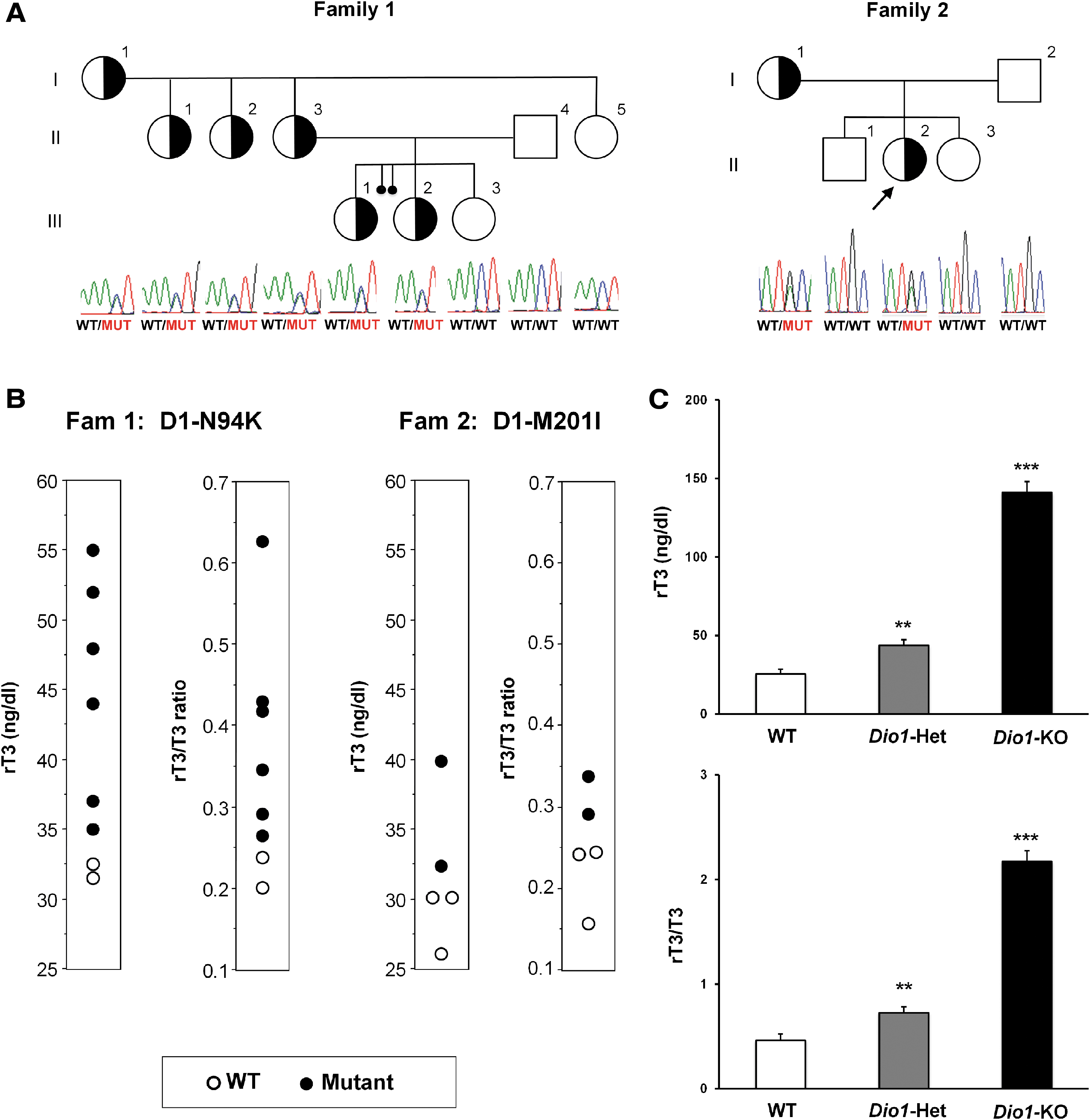
Pedigree of the families 1 and 2, genetic analysis, serum rT3 concentration, and rT3/T3 ratios in humans and Dio1-KO mice. (
Mouse models of D1 deficiency
Although no mutations in DIO1 gene have been previously reported in humans, a mouse with a natural deficiency of D1 (C3H/He strain) exhibited elevated serum rT3 level and rT3/T3 ratio (9,10), suggesting impaired D1-mediated rT3 clearance via outer ring deiodination. This was confirmed when the mouse model with targeted inactivation of the Dio1 gene was found to have elevated serum rT3 level and rT3/T3 ratio (6). Here, we studied heterozygous Dio1 knock-out mice backcrossed >10 times into the WT 57BL/J5 strain (5) for comparison to affected members in both families. Similar to the heterozygous humans, they had elevated serum rT3 levels (Dio1-Het 43.7 ± 3.5 vs. WT 25.3 ± 3.1 ng/dL, **p < 0.01) as well as elevated rT3/T3 ratios (Dio1-Het 0.725 ± 0.059 vs. WT 0.462 ± 0.062, **p < 0.01) (Fig. 1C).
Enzymatic activities
To assess the functional activity of the mutant D1 proteins (D1-N94K and D1-M201I), site-directed mutagenesis was used to obtain the pcDNA3-based vectors pD1-N94K or pD1-M201I from the pD1-WT, as detailed in the Materials and Methods section. Plasmids were transiently expressed in HEK-293 cells using pGFP as transfection control. D1 velocity was studied in cell sonicates in the presence of 0.1–10 μM 125IT4 and 10 mM DTT and was found to be reduced for both mutants compared with cells expressing pD1-WT (Fig. 2A, B). After data reduction through the Lineweaver–Burk plot, D1 kinetics revealed two- and threefold higher Km[T4] for pD1-N94K and pD1-M201I, compared with pD1-WT, 20.4 and 28.7 μM versus 9 and 11 μM, respectively. Deiodinases dimerize but may preserve two functionally independent active centers (11). To find out if the mutant D1 proteins could interfere with the catalytic activity of the WT D1, we used the HEK-293 in vitro cell system to co-express equal amounts of pD1-WT and pD1-N94K (pD1-WT/N94K) and compare with the enzymatic activity of pD1-WT or that of pD1-N94K (Fig. 2E); similar studies were performed for the pD1-M201I mutant (pD1-WT/M201I) (Fig. 2F). In each case, that is, the co-expression of pD1-WT and pD1-N94K or pD1-WT and pD1-M201I, the resulting D1 activity corresponded to approximately the average between the independently expressed WT and mutant D1 proteins (Fig. 2E, F). This pattern is suggestive of haploinsufficiency as an underlying mechanism (Fig. 2E, F), reminiscent of the haploinsufficiency effect in the mice Dio1-Het null versus WT and Dio1-KO mice in terms of serum thyroid abnormalities (Fig. 1C).
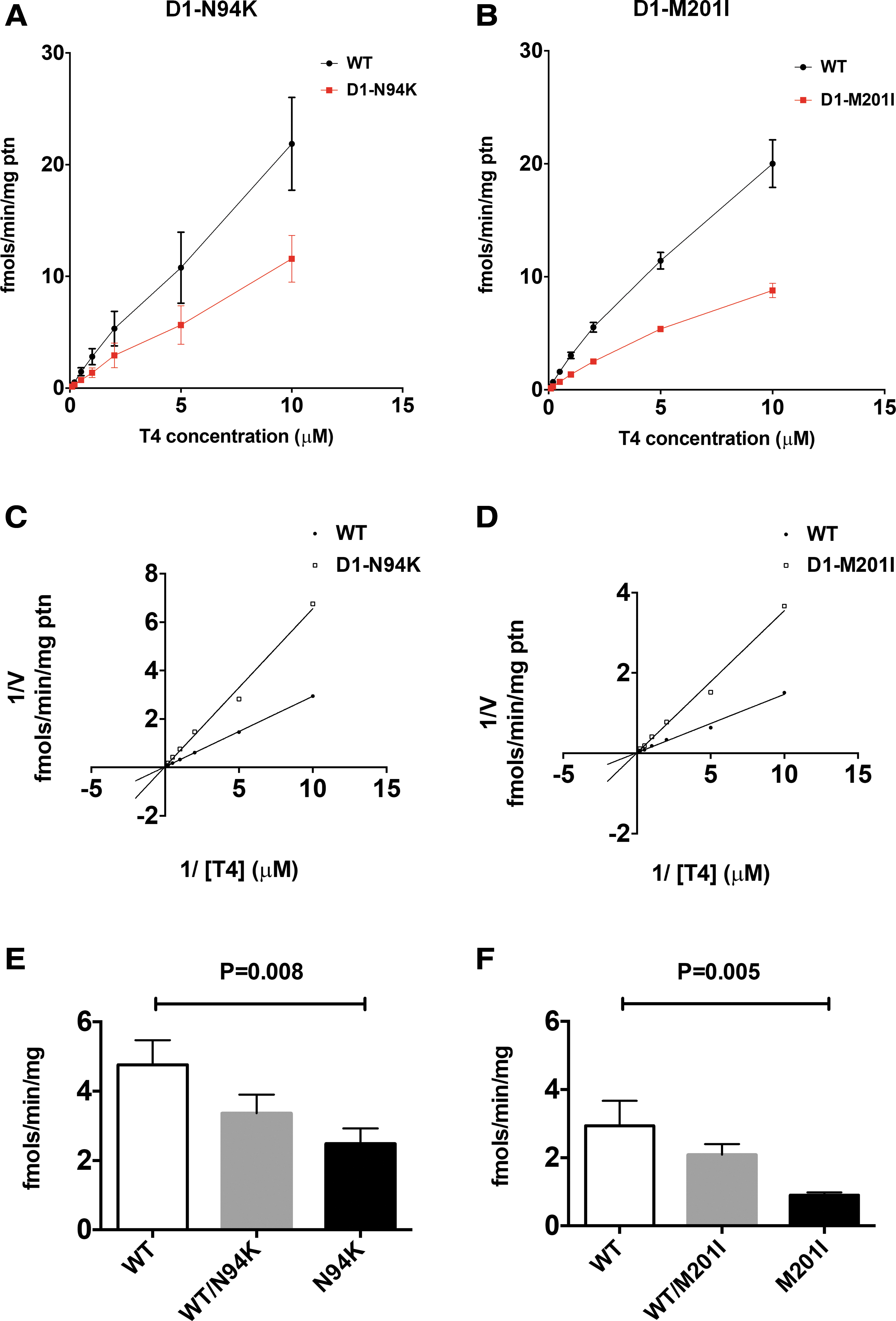
Kinetic analysis of D1 mutants. Deiodinase activities were measured in sonicated HEK-293 cells transiently expressing the indicated mutants. (
Discussion
The selenoenzyme deiodinases modulate the availability of the active TH, T3 (1). In particular, the D1 enzymatic activity modulates the serum levels of T3 largely due to its high expression in the liver and due to its relatively high Km for the substrate T4. Genetic defects in the human deiodinases genes have remained elusive. Dio1 deficiency in mice was reported to occur naturally in the strain CH3, and targeted genetically engineered Dio1-deficient mice have also been generated and both mouse models have elevated serum rT3 levels (9,10).
The identification of the pathogenic variants N94K and M201I in two unrelated families offered the opportunity to study the consequences of D1 deficiency in humans. Heterozygous affected in both families had elevated serum rT3 and rT3/T3 ratio, similar to the two mouse models. Overall, the clinical phenotype of the affected individuals is relatively mild with no obvious abnormalities other than serum thyroid tests. To assess for other potential altered functional parameters in these patients, we measured liver enzymes, total cholesterol, blood urea nitrogen, and creatinine in members of family 2 (samples from family 1 were not available for these studies). As shown in Supplementary Table S1, cholesterol shows very high level in the affected mother (I-1 in Fig. 1A, family 2) and upper normal level of 199 in the 23-year-old proposita (II-2), while her father (I-2) and brother (II-1) have normal levels. Thus, it is possible that elevated cholesterol could be another functional parameter in patients with DIO1 mutations, correlated with the decreased capacity to locally generate T3 in the liver. It remains unknown how does nonthyroidal illness manifest in them in setting of an already low D1 enzymatic activity. At the same token, the decreased capacity for the D1 enzymatic activity is expected to affect instances when increased availability of serum T3 is required. Further clinical evaluation of these patients over time will be needed to answer these questions.
The kinetic studies of the mutant D1 proteins demonstrate two to threefold higher Km indicating lower substrate affinity and slower enzyme velocity. Next, looking at the relevance of the two amino acids in the context of all three deiodinases, both N94 and M201 residues are highly conserved among D1, D2, and D3 in humans (12) and across species. One possible aspect to study in the future would be to generate the respective mutant D2 and D3 protein variants to assess the functional consequences for their enzymatic activity. To date, only D3 has been successfully crystalized (13), but without resolving the structure of active deiodinase crystals, it is difficult to speculate how these mutations could be affecting the enzyme's catalytic activity. M201 is located within the thioredoxin-like fold domain of D1, a structure that is shared by the three deiodinases (12). Because of its proximity to the selenocysteine (Sec)-containing catalytic active center, M201 could have a direct role in substrate binding or the deiodination reaction. At the same time, N94 is located in the hinge segment, connecting the transmembrane domain and the globular catalytic active center. This segment is known to play a role in deiodinase dimerization, a process that is critical for its catalytic activity (14).
An overall mechanism of haploinsufficiency is supported by the co-expression of WT D1 with either one of the mutants N94K or M201I. Resulting D1 activities correspond to approximately the average between the independently expressed WT and respective mutant D1 proteins. While the deiodinases dimerize, they may preserve two functionally independent active centers (11). This hypothesis of haploinsufficiency in D1 enzymatic capacity as the underlying mechanism is also supported by the similar elevation in rT3 and rT3/T3 ratio in affected individuals compared with the Dio1 heterozygous-null mice.
We report the identification and characterization of two missense DIO1 pathogenic variants in unrelated families presenting with abnormal TH metabolism, representing the first demonstration of inherited D1 deficiency in humans.
Footnotes
Acknowledgments
The authors thank the patients and their families for participating in this study.
Author Disclosure Statement
M.M.F., X.-H.L., G.W.F., A.G., S.R., and A.M.D. declare no competing financial interests. A.C.B. is a consultant for Allergan Inc., Synthonics Inc., and BLA Technologies LLC.
Funding Information
This work was supported by grants DK15070 to S.R., DK110322 to A.M.D., and DK58538 and DK65055 to A.C.B. from the National Institutes of Health, USA.
Supplementary Material
Supplementary Table S1