
Abstract
Background:
Lenvatinib, a multikinase inhibitor, is for progressive radioiodine-refractory-differentiated thyroid cancer (RR-DTC) patients. However, there are a lot of drug-related adverse events (AEs) that can affect the quality of life (QoL) of patients. The aims of this study were (a) to evaluate, and compared with other series, the safety of lenvatinib used in RR-DTC patients enrolled in an Italian expanded access program (EAP), and (b) to evaluate their QoL during treatment with lenvatinib.
Methods:
To evaluate the safety, we recorded and graded all AEs during the 6 months of lenvatinib treatment in 39 RR-DTC patients. We compared the safety profile of lenvatinib observed in our patients with that reported in the study of (E7080) levatinib in differentiated cancer of the thyroid (SELECT) and tumeurs thyroidiennes refractaires (TUTHYREF) network studies. Moreover, we evaluated the QoL in our series by using the European Organization for Research and Treatment (EORTC) Quality of Life Questionnaire-Core 30 and the pain visual analogue scale (VAS).
Results:
The most frequent AEs among our 39 RR-DTC patients were hypertension (80.5%), fatigue (58.3%), diarrhea (36.1%), stomatitis (33.3%), hand/foot syndrome (33.3%), and weight loss (30.5%). The most prevalent grade 3/4 AE was hypertension (25%). When compared with previous studies (i.e., SELECT and TUTHYREF), a significantly lower percentage of our patients experienced diarrhea, nausea, proteinuria, and weight loss. No statistically significant differences in the QoL of our patients evaluated before, during, and at the end of follow-up (6 months after starting the therapy) were found. However, a slight improvement of the general health and emotional and cognitive status associated with a slightly worsening of physical role and social functioning was observed during these 6 months. Pain, dyspnea, insomnia, and constipation moved toward better values, while fatigue, nausea and vomiting, appetite loss, and diarrhea worsened. By comparing the pain VAS, an overall reduction of the level of pain was found.
Conclusions:
The safety profile of the drug was similar to that already reported with some differences in the prevalence and severity of the AEs. Regarding the QoL, the EAP showed a trend of improvement of the global health status and a reduction of symptoms correlated to the disease. The clinical impact of fatigue, anorexia/weight loss and stomatitis, mainly due to the drug itself, continues to represent the major issue in the management of these patients.
Introduction
The initial treatment for advanced differentiated thyroid cancer (DTC) is total or near-total thyroidectomy, followed, whenever necessary, by radioactive iodine (131I) and thyroid hormone suppressive therapy (1). 131I avidity is associated with a favorable prognosis even in patients with recurrent or metastatic disease. However, some patients are resistant to 131I therapy, in which case the 10-year survival rate will be as low as <10% (2). Furthermore, cytotoxic chemotherapy is not very effective in patients with metastatic, rapidly progressive, symptomatic, and/or imminently life-threatening radioiodine-refractory (RR)-DTC (3). Alternatively, current advances in multikinase inhibitor (MKI)-targeted therapies have offered novel possibilities to manage DTC in these patients (4).
Lenvatinib is an oral MKI, which is effective against vascular endothelial growth factor receptors (1 –3), fibroblast growth factor receptors (1 –4), platelet-derived growth factor receptor alpha, and the rearranged during transfection (RET) and c-KIT proto-oncogenes (5,6). In the phase III study of (E7080) levatinib in differentiated cancer of the thyroid (SELECT) trial, lenvatinib was shown to significantly prolong the progression-free survival (PFS) in patients with DTC by almost 15 months compared with placebo (7). Similarly, a few other studies about the efficacy of lenvatinib in patients with RR-DTC confirmed a similar safety profile but a lower PFS in a real-world clinical setting (8 –11).
Lenvatinib was approved by the U.S. Food and Drug Administration and the European Medicine Agency (EMA) in February and May of 2015, respectively. This was followed by initiating an expanded access program (EAP) study in Italy whose aims were to assess (a) the safety outcomes and (b) the quality of life (QoL) of MKI-naive patients with RR-DTC following lenvatinib treatment. In this study, we analyzed the safety results and compared them with those obtained in the multicenter, international, randomized phase III clinical trial (SELECT study) (7) and in the first study collecting data from the real-life practice (tumeurs thyroidiennes refractaires [TUTHYREF] network study) (8). Moreover, data on QoL of patients treated with lenvatinib were prospectively collected for the first time and reported in the present study.
Materials and Methods
Study design
This was an observational, open-label, multicenter EAP study involving patients with RR-DTC at eight Italian sites over 14 months, between April 2015 and June 2016. The program consisted of two phases: pretreatment and treatment. The endpoints of this study were safety and QoL. Safety was evaluated by monitoring and recording all adverse events (AEs) and serious AEs. AEs were assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0 (12). Hematology, clinical chemistry, and urine values were systematically monitored. Physical examinations, electrocardiograms (ECGs), and regular measurement of vital signs were performed according to the schedule of visits and procedures. All patients had an echocardiogram during the pretreatment phase and whenever clinically indicated. Last, patients were invited to take note of the AE (i.e., type of AE, date of onset, severity/degree, duration) using a diary and to call immediately at the onset of any AE. A comparison of the safety profile of lenvatinib in our patients with that reported in SELECT and TUTHYREF network studies was performed (7,8).
QoL was assessed using the European Organization for Research and Treatment (EORTC) Quality of Life Questionnaire-Core 30 (QLQ-C30) model (13) and the pain visual analogue scale (VAS) (14). Patients were asked to self-complete the two questionnaires at screening, baseline, and then every 28 days for six cycles (i.e., 6 months from the initiation of therapy). The QoL EORTC QLQ-C30 was specifically designed for cancer patients (15). It consists of 30 questions and incorporates a Global Health Status/QoL scale, 5 functional scales (physical functioning [PF2]: 5 questions; role functioning [RF2]: 2 questions; cognitive functioning [CF]: 2 questions; emotional functioning [EF]: 4 questions; social functioning [SF]: 2 questions), and 9 symptom scales (fatigue, nausea and vomiting, pain, dyspnea, insomnia, loss of appetite, constipation, diarrhea and financial difficulties). EORTC QLQ-C30 scales are scored on a 0–100 scale: high scores on the functioning scales and on the global health/QoL scale indicate good QoL, while high scores on the symptom scales indicate reduced QoL (13). The pain VAS uses a scale from 0 to 100 mm, with 0 mm indicating no pain and 100 mm indicating the worst possible pain (14).
All patients provided written informed consent before starting the pretreatment phase, and the study protocol was approved by all relevant institutional ethics committees. The study was conducted in accordance with the provisions of the Declaration of Helsinki (2013) and after the approval of the local ethic committees.
Patients
Patients were eligible for enrollment if they had a histologically confirmed diagnosis of papillary thyroid cancer (PTC) or follicular thyroid cancer (FTC) and evidence of RR-DTC, according to at least one of the following criteria: one or more lesions that did not demonstrate 131I uptake on a radioiodine scan; one or more lesions that had substantially increased in size within 12 months of 131I-therapy, regardless of radioiodine activity during pre- or post-treatment; cumulative radioiodine activity of >600 mCi (22 gigabecquerels), with the last activity administered at least 6 months before program entry. Patients with poorly differentiated thyroid cancer (PDTC) and Hürthle carcinoma (HC) could be also enrolled. To be treated, patients were required to have a progressive disease in the last 12 months according to the Response Evaluation Criteria In Solid Tumors version 1.1 standards (16) and not to have received prior therapy with MKIs. Furthermore, patients had to have a good Eastern Cooperative Oncology Group (ECOG) performance status (0–2), blood pressure of ≤150/90 mmHg, and adequate liver, kidney, and bone marrow functions.
Conversely, patients with proteinuria ≥1 g/24 hours, history of congestive heart failure (New York Heart Association class >2), ECG with corrected QT interval ≥480 ms, and existing anticancer therapy-related toxicities of grade ≥2 (except alopecia and infertility) were excluded from the program.
Treatment
Lenvatinib was administered to patients at a starting dose of 24 mg (two 10 mg capsules and one 4 mg capsule) once daily (QD), until withdrawal of consent, disease progression, unacceptable toxicity, or death. Dosing reductions and treatment interruptions were permitted to manage treatment-related toxicity. Dose reductions occurred in succession based on the previous dose level (20, 14, and 10 mg/day), and once the dose was reduced, it was not increased again.
Statistical analyses
Continuous variables were presented as the mean ± standard deviation (SD) or median and interquartile range, while categorical variables were presented as frequencies and percentages. Fisher exact test or chi-squared test was used to measure the significance of categorical data.
QoL scores at baseline were summarized as mean ± SD and as median and minimum–maximum. Longitudinal linear models were estimated to depict any changes in each QoL score throughout the treatment period accounting for the repeated measures design using a spatial power covariance matrix. Results were reported as regression coefficients (mean change per 1 month of follow-up) along with standard error and p-value. Estimated means in each month of follow-up were used for graphical purposes. A p-value <0.05 was considered statistically significant. All statistical analyses were performed using SAS version 9.4 software (SAS Institute, Cary, NC).
Results
Clinical and pathological features at enrollment
Forty-one patients were screened during the EAP study pretreatment phase and 39 were included in the treatment phase. Two patients were excluded because the ECOG was >2. Baseline demographic and clinical characteristics are summarized in Table 1. Median age at enrollment was 64 years (range 45–80 years), with a higher rate of male patients; the most frequent histotype was PTC, which was present in 27 (69.2%) patients, followed by FTC in 14 (35.9%) patients (2 patients had both PTC and FTC). All patients underwent total thyroidectomy followed by 131I. At enrollment, the most frequent sites of metastases were lung, lymph node, bone, and liver with a prevalence of 83.8%, 75.7%, 40.5%, and 13.5%, respectively. ECOG was ≤1 in 34/39 (87.2%) patients and 2 in the remaining 12.8% of patients (Table 1). All patients had a progressive RR-DTC and were naive to MKI therapy.
Demographic and Clinical Characteristics of Patients at Enrollment in the Expanded Access Program
Two patients with both PTC and FTC.
ECOG/NYHA, Eastern Cooperative Oncology Group/New York Heart Association; FTC, follicular thyroid cancer; PTC, papillary thyroid cancer; SD, standard deviation.
Safety
Thirty-six of 39 patients were evaluable for toxicity. Data of toxicity of the other three patients were not available because they withdrew early the consent to study participation, although continuing the treatment for ethical reasons. During lenvatinib treatment, all 36 patients experienced at least one AE. Arterial hypertension was the most frequent AE, reported in 29 (80.5%) patients, starting after a median of 14 days from the beginning of therapy (Fig. 1). After hypertension, the most frequent AEs were fatigue in 21 (58.3%) patients after a median of 27 days of therapy; diarrhea in 13 (36.1%) patients after a median of 85 days of therapy; mucositis/stomatitis and hand/foot syndrome in 12 (33.3%) patients after a median of 33 and 80 days of therapy, respectively; weight loss in 11 (30.5%) patients early after the start of therapy, however, these patients developed the highest grade of this AE after a median of 83 days. A quarter of patients reported anorexia and dysphagia. Dysphonia, myalgia/arthritis, nausea, proteinuria, and skin rash were reported in 27.8%, 22.2%, 11.1%, 11.1%, and 8.3% of patients, respectively. Most patients had AEs of grade 1 or 2, while grade 3 or 4 AEs were reported in 19 (52.8%) patients. As shown in Table 2 the most frequent grade 3 and 4 AEs were hypertension (9/36 [25%]), mucositis/stomatitis (3/36 [8.3%]), and fatigue (3/36 [8.3%]). During treatment, most patients required a dose adjustment [reduction or temporary interruption (31/39; 79.5%)] due to AEs and the first dose reduction occurred after a median of 2 months from the start of therapy. The most frequent AEs leading to dose adjustment were mucositis/stomatitis, anorexia/dysphagia, and fatigue, all responded to dose reduction in terms of improvement of grade. After 3 months of therapy, the median daily dose of the drug was 14 mg per day and this was maintained until the end of the follow-up (6 months of therapy).

Prevalence and median time of appearance of the six most frequent lenvatinib-related AEs in the EAP study. AEs, adverse events; EAP, expanded access program.
Most Common Lenvatinib Treatment-Related Adverse Events: Degree of Severity and Time of Onset from Starting Therapy
CTCAE, Common Terminology Criteria for Adverse Events.
At present (i.e., 5 years after starting the EAP study), 11/39 (28.2%) patients are still on lenvatinib treatment. The overall median time of duration of treatment was 36 months (range 2–58 months). Patients still under treatment are taking either 14 or 10 mg per day of lenvatinib with a relatively good QoL.
Comparison of safety profiles between the EAP and other studies
The clinical and pathological features of patients in the EAP study were similar to those reported in other studies (7,8). Median age at enrollment, sex distribution, different histotypes, ECOG performance status, and sites of metastases are listed in Table 3. In the EAP study, only patients with PTC and FTC were recruited, while in the other studies, also PDTC and HC were enrolled. Limiting the comparison with PTC and FTC, the distribution of these two histotypes was similar across the other studies (Table 3).
Comparison of Pre-enrollment Features Between Expanded Access Program Group, Study of (E7080) Levatinib in Differentiated Cancer of the Thyroid Group, and Tumeurs Thyroidiennes Refractaires Network Study
p: EAP versus SELECT; p 1: EAP versus TUTHYREF network.
Two patients with both PTC and FTC.
In EAP study, the analysis was available in 37 patients; in TUTHYREF network study, the rate of metastases was calculated in 74 patients.
When considering only PTC and FTC histotypes.
When considering all histotypes.
EAP, expanded access program; ns, not statistically significant; SELECT, study of (E7080) levatinib in differentiated cancer of the thyroid; TUTHYREF, tumeurs thyroidiennes refractaires.
We examined the type, prevalence, and severity of AEs observed in the EAP study with those reported in the SELECT and TUTHYREF network studies (Table 4). Compared with the SELECT trial, we found a significantly lower proportion of patients who experienced diarrhea (36.1% vs. 59.4% in SELECT, p = 0.008), nausea (11.1% vs. 41% in SELECT, p = 0.0005), and proteinuria (11.1% vs. 31% in SELECT, p = 0.01). Compared with the TUTHYREF network study, the only difference was in weight loss, present at a lower percentage in our patients (30.5% vs. 59%). In contrast, a similar proportion was reported for arterial hypertension, fatigue, stomatitis, hand/foot syndrome, myalgia, and skin rash. Only minor differences were found in the severity of AEs between patients in the EAP study and the SELECT study (except for arterial hypertension, which was grade ≥3 in 31% of hypertensive patients vs. 61.6% of hypertensive patients in the SELECT study; p = 0.002; Table 4). However, a similar distribution in the severity of AEs was found between patients in the EAP and the TUTHYREF network study (Table 4). Overall, 79.5% of our patients required a dose reduction to manage drug-related AEs compared with 68% and 59% in the SELECT and TUTHYREF network study, respectively. However, in our study, only 20.5% of patients had treatment discontinuation (for any reason) compared with 42.7% in the TUTHYREF network study. No information about the percentage of discontinuation for any reason is provided in the SELECT study.
Comparison of Adverse Events Between Expanded Access Program Group, SELECT Group, and TUTHYREF Network Study
p: EAP versus SELECT; p 1: EAP versus TUTHYREF network.
Data derived from SELECT study analysis.
Data derived from TUTHYREF network study.
Quality of life
The EORTC QLQ-C30 and the VAS were correctly completed by 27 and 34 patients, respectively, during a median treatment and follow-up period of 5.4 months (range 0.5–7.8 months). The pain functional and symptom scales and the global health status scale were derived from raw data and their baseline values are reported in Table 5.
Quality-of-Life Score for Functioning and General Symptom Dimensions of Radioiodine-Refractory Differentiated Thyroid Cancer Patients During Lenvatinib Therapy
A p-value <0.05 was considered statistically significant.
QoL, quality of life; SD, standard error.
By comparing the EORTC QLQ-C30 at the time of enrollment and during the treatment phase, we found that the median global health status (QL2) increased by 2.56 points from baseline to 6 months of therapy (Supplementary Fig. S1, panel a), however, this was not statistically significant (p = 0.608, Table 5).
EF and CF increased over the 6 months (Supplementary Fig. S1, panels d and e), however, this difference was not statistically significant (Table 5). Conversely, the PF2, role functioning, and SF deteriorated during lenvatinib treatment (Supplementary Fig. S1, panels b, c, and f), but this was also not statistically significant (Table 5). Furthermore, we observed that some symptom subscales improved, such as pain, dyspnea, insomnia, and constipation (Supplementary Fig. S1, panels i–k and m, respectively); conversely, others such as fatigue, nausea/vomiting, appetite loss, diarrhea, and financial difficulties worsened (Supplementary Fig. S1, panels g, h, l, n, and o, respectively). Despite these minor differences, no significant variations were observed in the improvement or deterioration of symptoms, except for diarrhea, which significantly worsened (p = 0.01) during the 6 months of lenvatinib treatment (Table 5), showing an increase of 13.5 points from baseline to 6 months of therapy (Supplementary Fig. S1, panel n).
By comparing the pain VAS at the time of enrollment and at the last control, we observed that there was an overall reduction in the level of pain (Fig. 2), although not statistically significant (p = 0.277) (Table 5). When we analyzed the results of the VAS in the entire group of patients, we found that 31% declared an improvement in pain, 50% did not report any worsening or improvement of pain, and only 19% complained of worsening pain.
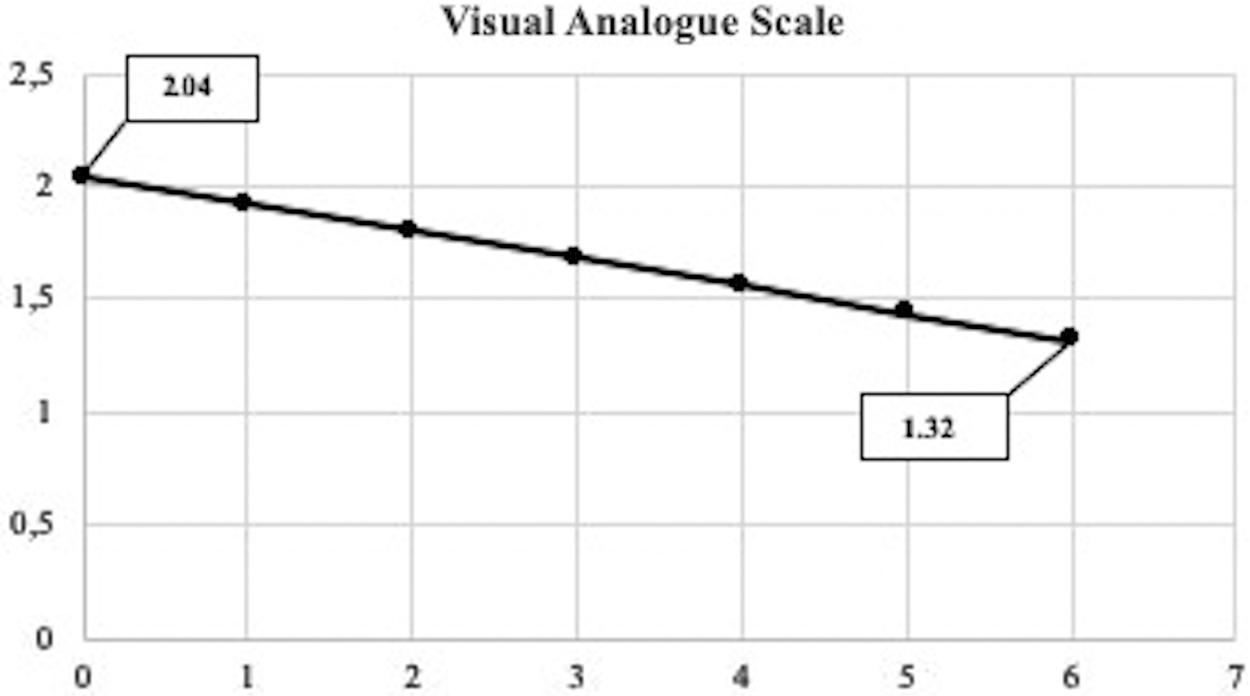
Mean visual analogue scale scores for pain, from baseline to 6 months of therapy. On the x-axis is represented the time in months, and the y-axis shows the estimated mean score of pain.
Discussion
Lenvatinib has been approved for the treatment of RR-DTC and much data have been collected from the phase III SELECT study (7). To date, no other clinical trials have reported results, and data from an ongoing comparative study using two different starting doses of lenvatinib (
In contrast to the SELECT study, the primary objective of the EAP study was to evaluate the safety profile and the QoL in patients treated with lenvatinib. The rationale was to asses if the clinicians could improve their ability to manage lenvatinib and maintain the patients under therapy. In a post hoc analysis performed on patients enrolled in the SELECT study, it was demonstrated that higher rates of dose interruption and/or dose reduction had a negative impact on the PFS (17). We observed a higher percentage of patients who had a dose interruption/reduction than in the SELECT and the TUTHYREF network studies (7,8). However, we had a much lower number of patients who totally discontinued the drug, for any reason, compared with the TUTHYREF network study (no comparison could be done with the SELECT study). This finding can be explained by the fact that in our centers there was a greater propensity to interrupt or reduce the drug as soon as possible after the development of an AE, with the aim to restart and continue the treatment as long as possible. The majority of our patients who interrupted/reduced the drug restarted the therapy as demonstrated by the few patients who definitively discontinued the drug during the 6 months of follow-up.
Interestingly, we found that the type of AEs was the same as already reported in other studies, but the frequency differed since severe hypertension (grade 3) was much more frequent in the other studies (7,8) compared with ours. Also, other AEs, especially diarrhea, nausea, and proteinuria, were less frequent in our patients than in those of the SELECT study, and weight loss was less frequent than in the TUTHYREF network study. These differences could be, at least in part, explained by the lower median daily dose of the drug in our study (14 mg) compared with the median dose of the TUTHYREF network study (20 mg) and the mean dose of the SELECT study (17.2 mg). In our clinical practice experience, even a small reduction of the daily dose (i.e., 4 mg daily) can be effective in reducing the severity of an AE and make the patient more adherent to therapy. The overall median duration of treatment in our series (36 months at the present) is longer than that observed in the SELECT study (19.4 months) even when the data cutoff was updated to September 2016 (5 years after starting the study) (18). This difference might be explained by a substantially different way to manage the drug in respect to the severity of AEs, since we usually reduced the daily dose at the appearance of a grade 2 AE and this likely allowed patients to stay longer on drug treatment. Another possible explanation is that all patients enrolled in the EAP study were MKI naive, as opposed to SELECT in which prior therapy was allowed.
The second objective of the EAP study was the prospective analysis of QoL in RR-DTC patients treated with lenvatinib. To our knowledge, there is only one retrospective study reporting information about QoL in RR-DTC patients, in which only one type of questionnaire (i.e., linear analogue scale assessment) was used (19). Consistent with this study findings, we did not find any statistically significant difference in the QoL of patients before, during, and at the end of follow-up, although the results of the EORTC QLQ-C30 show a trend of improvement in QL2. This improvement was observed when the emotional and cognitive status was considered, while the physical role and SF were slightly deteriorated. An explanation for these contrasting results is the possibility that the enrollment into a clinical trial could be per se responsible for the improvement of the emotional and cognitive status of the patients who are approaching a new therapeutic opportunity with a lot of hope. Conversely, the AEs related to the drug itself could be responsible for the slight deteriorations in the patients' physical condition and limitation in their social life. It is conceivable that severe diarrhea can be a major limiting factor to participating in social activities; diarrhea was the only AE that significantly worsened, continued for many weeks, and reached its highest prevalence 2.8 months after the start of therapy. Regarding other symptoms, lenvatinib improved pain and dyspnea scores, likely because they are secondary to the disease, while those AEs due to the drug itself, such as fatigue, nausea, vomiting, appetite loss, and diarrhea, simultaneously worsened. Notably, pain scores, which can have a big impact on the QoL during long-term treatments, were improved or stabilized in 81% of patients.
In our opinion, the results of the present study indicate that to obtain a greater benefit from the therapy, the control and prevention of AEs are important since this allows treatment continuation at a lower daily dose but without interruptions that have been demonstrated to affect the efficacy of lenvatinib (17). Patients must be instructed and encouraged to report AEs as soon as possible, which would allow doctors to either prescribe adequate medications to help reduce/avoid AEs, or to proceed with an immediate dose reduction(s) to strike a healthy balance between tumor control and AE occurrence.
Our study has a major limitation due to the small number of patients, even if comparable with that of the already published study (n = 25) (19), who correctly completed the EORTC QLQ-C30 (n = 27) and the VAS (n = 34). The number of patients was limited by the nature of the study, since it was a study approved to make a temporary “bridge” between the approval of the drug by the EMA and the Agenzia Italiana del Farmaco. When lenvatinib was approved in Italy (15 months after the approval by EMA), the EAP study was over. Despite the small number of patients, our rate of enrollment was greater than that of the SELECT study as demonstrated by the fact that we enrolled 39 patients at 8 centers (4.8 patients per center) over 15 months, while in the SELECT study, 392 patients were enrolled at 117 centers over 14 months (3.35 patients per center).
The low number of correctly completed QoL questionnaires was due to the inexperience of some centers participating in the EAP study since, when the investigators collected the questionnaires, they did not verify that patients who should fill the forms by themselves without the influence of doctors or nurses did so. To avoid any misleading information, we discarded those questionnaires that were not appropriately completed. The low number of evaluated cases can be responsible for the lack of statistical significant differences. Although we would have preferred performing a QoL analysis in a larger population, the information we gathered from this small sample is useful, especially when considering the severity of the disease and the AEs that occurred, and how the antitumoral activity of the drug limited some symptoms related to the disease while giving rise to other symptoms due to the AEs of the drug itself.
Another limitation is that the response to therapy (i.e., the efficacy of the drug) was not an objective of our study and we could not compare the improvement or worsening of QoL with the response to treatment, which certainly can play a role in the perception and endurance of some symptoms both due to the disease and drug-related AEs by patients. However, we feel that, as for many other studies on the QoL of patients treated with new drugs or new chemotherapeutic schemes (20 –22) independent of the efficacy results, our study provides useful information, especially for colleagues who are not familiar with the use of lenvatinib.
In conclusion, this study showed that despite the increased experience of doctors in managing lenvatinib and its AEs, the safety profile of the drug was similar to that of the SELECT study, with minor differences observed in the prevalence and severity of AEs, while the types of AEs were similar to those of the SELECT and TUTHYREF network studies. Moreover, although in a limited number of cases, the EAP showed for the first time that there was a trend of improvement of the global health status of treated patients and, in particular, a reduction of symptoms correlated to the disease such as pain that worsened only in a minority (<20%) of patients. The problem of AEs, such as fatigue, anorexia/weight loss, and stomatitis, that are strictly correlated to some undesirable drug-related activities continues to represent the major issue in the management of these patients, and should be detected as soon as possible to avoid the drug dose interruption that can affect the efficacy of the drug (17).
Footnotes
Acknowledgment
C.G. contributed to this article as a recipient of the PhD program in Clinical and Translational Science of the University of Pisa.
Author Disclosure Statement
D.C., I.T., M.S., P.G., S.D., L.L., M.E., and E.R. are consultants for EISAI, however, the present study was not conditioned by this activity of these authors. G.C., V.L., B.A., G.G., M.M., P.F., P.T., T.M., and T.M. have nothing to disclose.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1