
Abstract
We investigated the genetic cause of thyroid dyshormonogenesis in a girl with congenital hypothyroidism. Genetic analysis showed that she was homozygous for a hitherto not described mutation (c.1432_1433delGT, p.V478KfsX11) in the solute carrier family 26 member 7 (SLC26A7) gene. SLC26A7 is proposed to be an anion transporter in the thyroid gland. The mutation leads to a frameshift and a premature stop codon. The predicted protein is truncated and very likely to be nonfunctional if it was expressed at all. In addition, in silico studies predict the mutation to be pathogenic.
Introduction
Approximately 15
Materials and Methods
Patient
The patient was the first daughter of consanguineous parents originating from Iran. She was born at term after an unremarkable pregnancy and raised in Germany in an area with moderate iodine deficiency. Neonatal screening was positive for congenital hypothyroidism that was confirmed at day 4 after birth (thyrotropin 704 mU/L, normal 0.2–4.2 mU/L, free T4 0.3 ng/dL, normal 0.9–1.7 ng/dL, free T3 1.8 pg/mL, normal 2.3–4.9 pg/mL). A defect in thyroid hormone synthesis was suspected because of a highly elevated serum thyroglobulin level (>3.000 ng/mL, reference range <50 ng/mL) and a goiter. This was confirmed by ultrasonography that also showed a highly increased vascularity.
On physical examination, she had jaundice and was hypotonic but otherwise normal. Treatment with
DNA amplification, sequencing, and in silico analysis
Clinical and genetic studies were approved by the institutional review board of the Johannes Gutenberg University of Mainz. Written informed consent was obtained from all family members to participate in this study. The coding sequence of the SLC26A7 gene was amplified by polymerase chain reaction (PCR) using genomic DNA. The methods for DNA extraction, PCR conditions, primer sequences, and details of the Sanger sequencing procedure are available upon request and will be provided by the authors. In silico analysis was performed utilizing GenomAD, EVS, dbSNP, Ensembl, UCSC genome browser, 1000genomes, and ClinVar.
Results
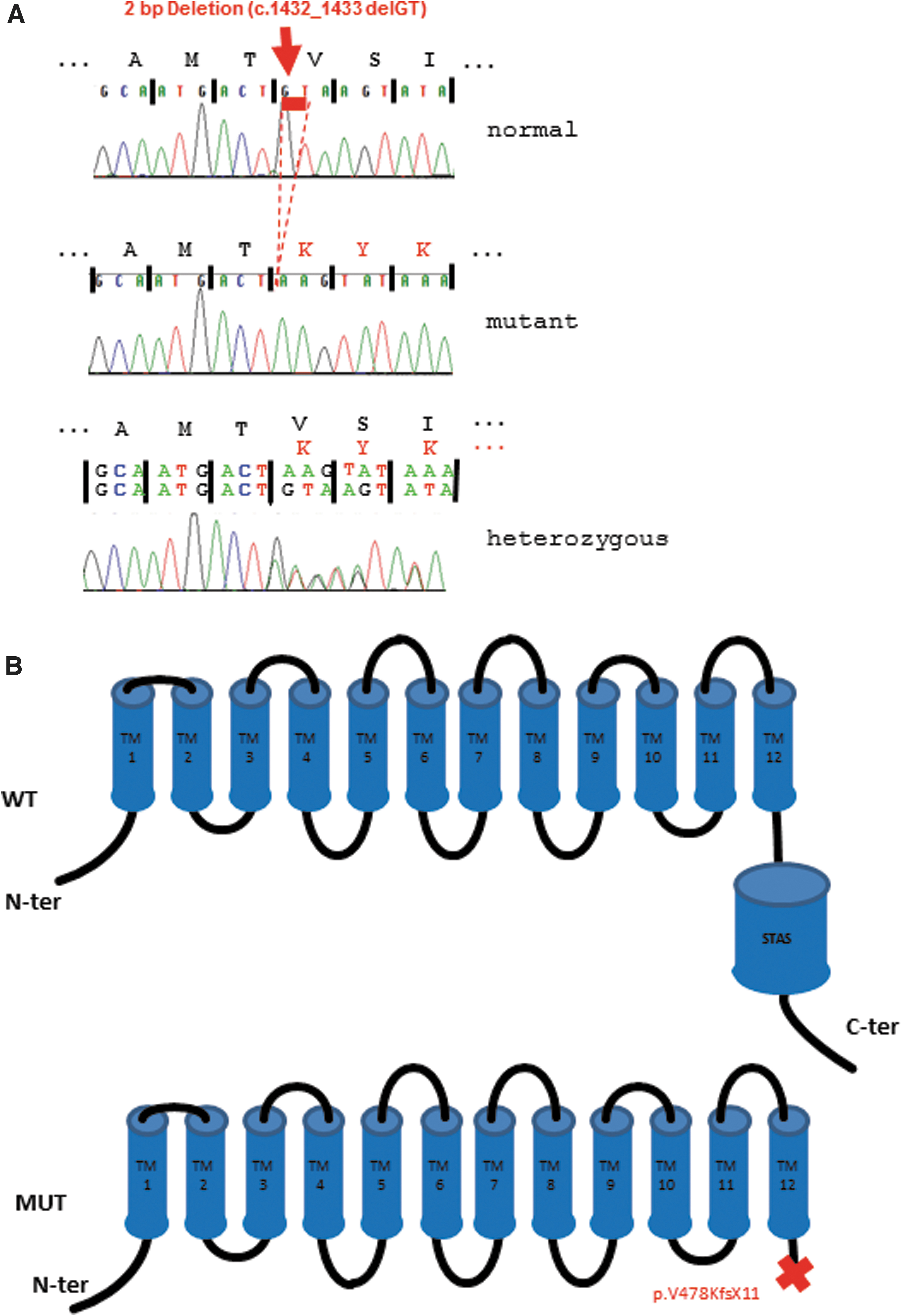
Sequencing analysis revealed a novel homozygous deletion (c.1432_1433delGT, p.V478KfsX11) in exon 14 of the SLC26A7 gene, which leads to a frameshift and a premature stop at position 489. Both parents and the brother were heterozygous carriers of this mutation (Fig. 1A). In silico analysis showed that c.1432_1433delGT, p.V478KfsX11 is not a common polymorphism and, therefore, pathogenic.
(
Discussion
We describe a rare case of thyroid dyshormonogenesis due to a novel homozygous mutation in the SLC26A7 gene. So far, only six SLC26A7 mutations have been identified (2 –5) that are nucleotide substitutions, deletions, or insertions leading to a truncated protein. We found two base pair deletions causing a frameshift and also a premature termination codon. The resulting transcript, if expressed at all, is predicted to translate into a severely truncated protein (Fig. 1B), which is not likely to be functional. Even though the exact location and function of SLC26A7 remain controversial (2,3), based on the studies of Ishii et al. it can be predicted that the truncation causes loss of the essential sulphate transporter and anti-sigma factor antagonist domain, which presumably leads to abnormal processing of the protein (3).
Interestingly, the phenotype of patients harboring SLC26A7 mutations varies. The majority of patients are diagnosed by neonatal screening and present with congenital hypothyroidism, goiter, and elevated serum thyroglobulin levels (2 –5), but hypothyroidism may also become evident later in life (3). Awareness of this condition will help to identify more patients harboring SLC26A7 mutations and eventually initiate further studies to elucidate the precise location and function of this protein.
Footnotes
Acknowledgement
Results shown in this article are part of the medical thesis of C.C.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding has been received for this research project.