
Abstract
Background:
BRAFV600E acts as an ATP-dependent cytosolic kinase. BRAFV600E inhibitors are widely available, but resistance to them is widely reported in the clinic. Lipid metabolism (fatty acids) is fundamental for energy and to control cell stress. Whether and how BRAFV600E impacts lipid metabolism regulation in papillary thyroid carcinoma (PTC) is still unknown. Acetyl-CoA carboxylase (ACC) is a rate-limiting enzyme for de novo lipid synthesis and inhibition of fatty acid oxidation (FAO). ACC1 and ACC2 genes encode distinct isoforms of ACC. The aim of our study was to determine the relationship between BRAFV600E and ACC in PTC.
Methods:
We performed RNA-seq and DNA copy number analyses in PTC and normal thyroid (NT) in The Cancer Genome Atlas samples. Validations were performed by using assays on PTC-derived cell lines of differing BRAF status and a xenograft mouse model derived from a heterozygous BRAFWT/V600E PTC-derived cell line with knockdown (sh) of ACC1 or ACC2.
Results:
ACC2 mRNA expression was significantly downregulated in BRAFV600E -PTC vs. BRAFWT -PTC or NT clinical samples. ACC2 protein levels were downregulated in BRAFV600E -PTC cell lines vs. the BRAFWT/WT PTC cell line. Vemurafenib increased ACC2 (and to a lesser extent ACC1) mRNA levels in PTC-derived cell lines in a BRAFV600E allelic dose-dependent manner. BRAFV600E inhibition increased de novo lipid synthesis rates, and decreased FAO due to oxygen consumption rate (OCR), and extracellular acidification rate (ECAR), after addition of palmitate. Only shACC2 significantly increased OCR rates due to FAO, while it decreased ECAR in BRAFV600E PTC-derived cells vs. controls. BRAFV600E inhibition synergized with shACC2 to increase intracellular reactive oxygen species production, leading to increased cell proliferation and, ultimately, vemurafenib resistance. Mice implanted with a BRAFWT/V600E PTC-derived cell line with shACC2 showed significantly increased tumor growth after vemurafenib treatment, while vehicle-treated controls, or shGFP control cells treated with vemurafenib showed stable tumor growth.
Conclusions:
These findings suggest a potential link between BRAFV600E and lipid metabolism regulation in PTC. BRAFV600E downregulates ACC2 levels, which deregulates de novo lipid synthesis, FAO due to OCR, and ECAR rates. ShACC2 may contribute to vemurafenib resistance and increased tumor growth. ACC2 rescue may represent a novel molecular strategy for overcoming resistance to BRAFV600E inhibitors in refractory PTC.
Introduction
The lack of effective treatments for aggressive papillary thyroid carcinoma (PTC) represents an unmet clinical need, especially given its rising incidence (1). PTC is the histological type of cancer with the highest frequency (2), and BRAFV600E is the most prevalent mutation driving cancer initiation and progression of PTC (3), while also correlating with increased mortality risk (4) and more aggressive clinic-pathological features with a higher probability of standard therapy failure (5). In 2011, the US Food and Drug Administration approved the first BRAFV600E inhibitor, vemurafenib, but unfortunately, resistance to this targeted therapy is widely reported in patients who initially responded (6). Our understanding of the molecular processes that allow BRAFV600E -positive tumors (including melanoma, colorectal adenocarcinoma, PTC, anaplastic thyroid carcinoma [ATC]) to develop resistance to BRAFV600E inhibitors (including vemurafenib) has advanced in recent years, and many studies have described several mechanisms: over-activation of the PI3K-AKT pathway due to increased c-MET expression (7); invasive processes resulting from epithelial-to-mesenchymal transition (8); focal amplifications of chromosome 6, with a minimal region of overlap that includes Met (9); aberrant HER3 overexpression mediated by autocrine loop signaling (10); autocrine interleukin-6 secretion via JAK/STAT3 and the MAPK pathway (11); persistent formation of the eIF4F complex (12); persistent activation of mTOR and hyperactive ERK signaling (13); expression of the BRAFV600E splicing isoform (14); BRAFV600E/K amplifications and non-MAPK alterations (15); ER stress response–mediated autophagy (16); copy number gain of MCL1 and loss of CDKN2A (P16) (17), also showing that CDK4/6 inhibitors, which mimic P16 function, in combination with vemurafenib induced apoptosis in PTC and ATC cells (18); and tetraploidization and amplification of chromosome 5 via mutations in the RNA-binding motifs (RBM) genes family (i.e., RBMX, RBM10) (18). Further, our recent studies of the tumor microenvironment showed that pericytes play an essential role in protecting human thyroid cancer cells from vemurafenib, sorafenib (a tyrosine kinase inhibitor), and combination therapy via TSP-1/TGFβ1 axis (19).
Researchers have begun to devote greater attention to deregulations in cell metabolism, among other hallmarks of cancer (20), to better understand the complexity of tumor biology, and several studies have already shed light on crucial metabolic aspects of various neoplasias (21). However, thyroid cancer metabolism remains poorly characterized, with very few studies focusing on the aerobic glycolytic phenotype or the role(s) of glutamine/amino acids (8) or the metabolic switch to glycolysis driven by PI3K (22). Moreover, lipid metabolism is fundamental for energy and makes available the reducing equivalents that control intracellular (23 –26) and microenvironment (27) stress, which may lead to tumor survival (28). Intriguingly, obesity (a multifactorial metabolic disease) is positively associated with greater frequency and higher mortality in thyroid cancer (1,29), but the biological mechanisms underlying these outcomes are completely unknown. One study reported lower lipid levels in thyroid lesions compared with normal thyroid (NT) tissue (30), but it did not detail tumor histotype or genetic events.
Whether and how the BRAFV600E oncogene impacts lipid metabolism regulation in thyroid cancer remains unknown. Acetyl-CoA carboxylase (ACC) is the rate-limiting enzyme that catalyzes the first committed step in fatty acid synthesis (FAS, which yields derived lipids), the ATP-dependent carboxylation of acetyl-CoA, which produces malonyl-CoA (31,32). Malonyl-CoA is the substrate for fatty acid synthase and is also an allosteric inhibitor of CPT1, the fatty acid transporter in the mitochondria that initiates fatty acid oxidation (FAO) (33); therefore, it is also crucial in regulating FAO. Humans (34) and other mammals have two paralogous genes encoding ACC: ACACA (ACCα or ACC1) and ACACB (ACCβ or ACC2), which are located on different chromosomes (17 and 12, respectively) and are believed to have originated from an ancestral gene duplication (33). Both ACCs belong to the biotin carboxylase protein family, and their enzymatic structure is well conserved, although they differ in N-terminal sequence (33), tissue-specific expression (34), and biological functions (35). ACC1 is primarily expressed in lipogenic tissue, such as liver and adipose (34), and produces malonyl-CoA which is directed to FAS; while ACC2 is prevalent in the heart and skeletal muscle (34), and it inhibits FAO. ACC2 is localized at the outer membrane of mitochondria (36), via its N-terminal sequence (37), while ACC1 is cytosolic. Indeed, the putative role of ACC2 is the inhibition of CPT1 (35), and it therefore performs a different function from ACC1. ACC1 and ACC2 also share post-translational modulation by phosphorylation and subsequent inactivation in two critical serine residues (Ser78 and Ser80 of human ACC1, Ser219 Ser221 of human ACC2) (38,39). AMP-activated kinase (AMPK), a well-described regulator, phosphorylates both ACC proteins (40), and is upstream-regulated by LKB1. Although the LKB1-AMPK axis has been generally described as tumor suppressive (41), AMPK activity as an energy sensor has conversely been shown to be useful to cancer cells in detecting metabolic stress (25). Transcription of ACC genes is mainly regulated by the sterol regulatory element-binding protein (SREBP-1) (42,43), which belongs to a family of helix-loop-helix-leucine zipper transcription factors. Increased de novo lipogenesis and reduced FAO may disturb different metabolic mechanisms and contribute to obesity. ACCs are considered drug targets for obesity, diabetes, and metabolic syndromes (44,45). Their modulation in tumors is more complex: ACC1 is upregulated in different tumor types (46,47), including nonsmall cell lung cancer (48). Conversely, ACC2 expression and/or activation are inhibited in tumors (49,50). ACC2 is stabilized and activated by PHD3 proline hydroxylation. In acute myeloid leukemia, PHD3 is significantly downregulated, which limits ACC2 activity (50). Further, on acidosis, cancer cells activate both FAS and FAO, selectively inhibiting ACC2 by epigenetic suppression (histone deacetylation) of the ACC2 promoter, which results in a direct proliferative advantage for tumor cells (49). Overall, these findings indicate that ACC1 and ACC2 have diverse roles in cancer metabolism, and only ACC1 inhibition is desirable as a drug target (51). However, dual ACC1 and ACC2 inhibitors have shown promising results in nonsmall-cell lung carcinoma preclinical models (52), and they have been tested in the treatment of hepatic steatosis (53).
Here, we assessed for the first time the role of ACC1 and ACC2 in PTC of differing BRAF mutation status, and their respective responses to therapy with vemurafenib both in vitro and in vivo. We have shown that ACC2 molecular action may contribute to resistance to BRAFV600E inhibition by modulating both de novo lipid synthesis and FAO, while also modulating the rate of intracellular oxidative stress in PTC-derived cell lines. ACC2, therefore, represents a novel target in the ongoing elucidation of the mechanisms underlying drug resistance for thyroid tumor progression.
Materials and Methods
Details regarding The Cancer Genome Atlas (TCGA) analyses, antibodies, cell cultures, vemurafenib treatment, real-time polymerase chain reaction, primers sequence, protein assays, cell proliferation, shRNAs, lipid assays, intracellular reactive oxygen species (ROS) assay, transmission electron microscopy, mouse model (mouse protocol was approved and performed in accordance with federal, local, and institutional guidelines at Beth Israel Deaconess Medical Center [BIDMC], Boston), and ultrasound can be found in Supplementary Data. The use of the following validated human PTC-derived cell lines (also defined here as PTC-derived cells) was approved by the COMS (BIDMC, Boston, MA): BCPAP is homozygous BRAFV600E (defined here as BRAFV600E/− ), KTC1 is heterozygous BRAFWT/V600E , and TPC1 has BRAFWT/WT and RET/PTC1 translocation (54).
Statistical analyses
Statistical analysis was carried out by using GraphPad Prism 7 software and Microsoft Excel. Chi-square test, t-student, Fisher's exact test, Mann–Whitney U test, one-way analysis of variance for multiple comparisons tests, and Pearson correlation analysis were used. Data are reported as the averaged value, and error bars represent the standard deviation or standard error of the mean for each group. Results with p-values below 0.05 were considered statistically significant.
Results
ACC1 and ACC2 mRNA expression levels in PTC and NT tissue samples
To determine ACC1 and ACC2 expression in PTC and NT clinical samples, we analyzed RNA-expression data from primary human thyroid tumors TCGA and metastasis, dividing the samples into four groups: BRAFWT/V600E -PTC, BRAFWT/WT -PTC, NT tissues, and metastasis. ACC1 mRNA levels were slightly higher on average in BRAFWT -PTC compared with the other groups, but the difference was very small and its expression levels were similar in BRAFV600E -PTC and NT (Fig. 1A). We also compared ACC1 RNA expression levels with other mutations occurring in PTC, to see whether genetic events other than the BRAFV600E mutation might have modulated its transcription levels; no significant associations were found (Fig. 1A). We then investigated whether histology type (classical, tall cell variant, or follicular variant), tumor size, sex, or age were associated with ACC1 expression levels, but we found no statistically significant differences (Fig. 1A). However, ACC1 expression levels were significantly different by BRAFWT and specific tumor stages (Fig. 1A) and also negatively correlated with the quantity (%) of enriched tumor stroma (r = −0.12123, p < 0.01). We next asked whether mRNA expression levels could be influenced by potential DNA copy number variations (CNVs) (Fig. 1B). We found a few copy number gains in BRAFWT -PTC, which positively correlated with ACC1 RNA expression levels (r = 0.2755, p < 0.001), and we observed no significant correlations in BRAFV600E-PTC and NT. We performed the same analysis for ACC2 mRNA expression levels and found that ACC2 levels were two-fold lower in BRAFV600E -PTC compared with BRAFWT -PTC, approximately four-fold compared with NT, and a two-fold difference between BRAFWT PTC and NT (Fig. 1C). Some tumor histological types appeared to be associated with the downregulation of ACC2; however, this association was highly confounded by the presence of BRAFV600E (Fig. 1C), as well as tumor stage. We estimated DNA CNVs (Fig. 1D) and as with ACC1, BRAFWT -PTC showed a few DNA copy number gains that correlated with ACC2 mRNA expression levels (r = 0.45, p < 0.001). We also checked whether age, sex, and tumor size were associated with ACC2 mRNA levels, but none of these factors showed any statistical significance. Finally, we found a negative correlation between ACC2 RNA expression levels and the quantity (%) of enriched tumors (r = −0.14807, p < 0.001) in PTC.

ACC1 (ACACA) and ACC2 (ACACB) gene expression in TCGA human PTC and NT tissue samples. (

New experimental model based on ACC expression in human PTC-derived cell lines
We next wanted to see whether our TCGA analysis was consistent with our in vitro models of PTC (Fig. 2A). We first analyzed ACC1 and ACC2 protein expression at baseline, and we saw that both BRAFV600E PTC-derived cell lines had lower levels of ACC2 compared with BRAFWT/WT ; more importantly, these ACC2 decreases looked to be dependent on gene dosage of BRAFV600E (Fig. 2B). Indeed, the BRAFV600/− PTC-derived cell line consistently had less ACC2 protein expression than BRAFWT/V600E . We further analyzed the phosphorylation status of ACC1, ACC2, and AMPK, since post-translational modification is linked to inactivation of these two enzymes, and AMPK is the best-characterized kinase targeting ACC. We found that both heterozygous and homozygous BRAFV600E PTC-derived cell lines have significantly higher ACC phosphorylation levels (1.6- and 4-fold changes, respectively) than the BRAFWT/WT PTC-derived cell line (Fig. 2B–D). In addition, AMPK phosphorylation levels rose 1.3-fold in the heterozygous BRAFV600E PTC-derived cell line only. Due to indistinguishable epitope recognition by pACC antibody on ACC1 versus ACC2 proteins, we were not able to precisely determine which isoform is predominantly phosphorylated in thyroid cancer cells. Thus, we performed selective immunoprecipitation (IP) for ACC1, ACC2, and pACC (Fig. 2E) to first assess each protein phosphorylation level at baseline, and second, since ACC2 produced two close bands in the Western blots (Fig. 2B) [the only known splicing variant is described in adipose tissue (55)], to assess the specificity of the two bands. ACC1 was consistently phosphorylated in all three PTC-derived cell lines (Fig. 2E), while ACC2 was phosphorylated only in the BRAFV600E PTC-derived cell lines, but at a substantially lower rate than ACC1 (Fig. 2E), suggesting that BRAFV600E -positive thyroid tumor cells post-translationally deregulate ACC1 and ACC2 and render them inactive. ACC2 protein expression showed different intensity in the two bands in the IP versus the specific input, which may have been caused by co-migration after the IP process. Further studies will be needed to interpret the biological meaning of the two bands; for all subsequent analyses we have quantified the intensity of both bands, since both were modulated.
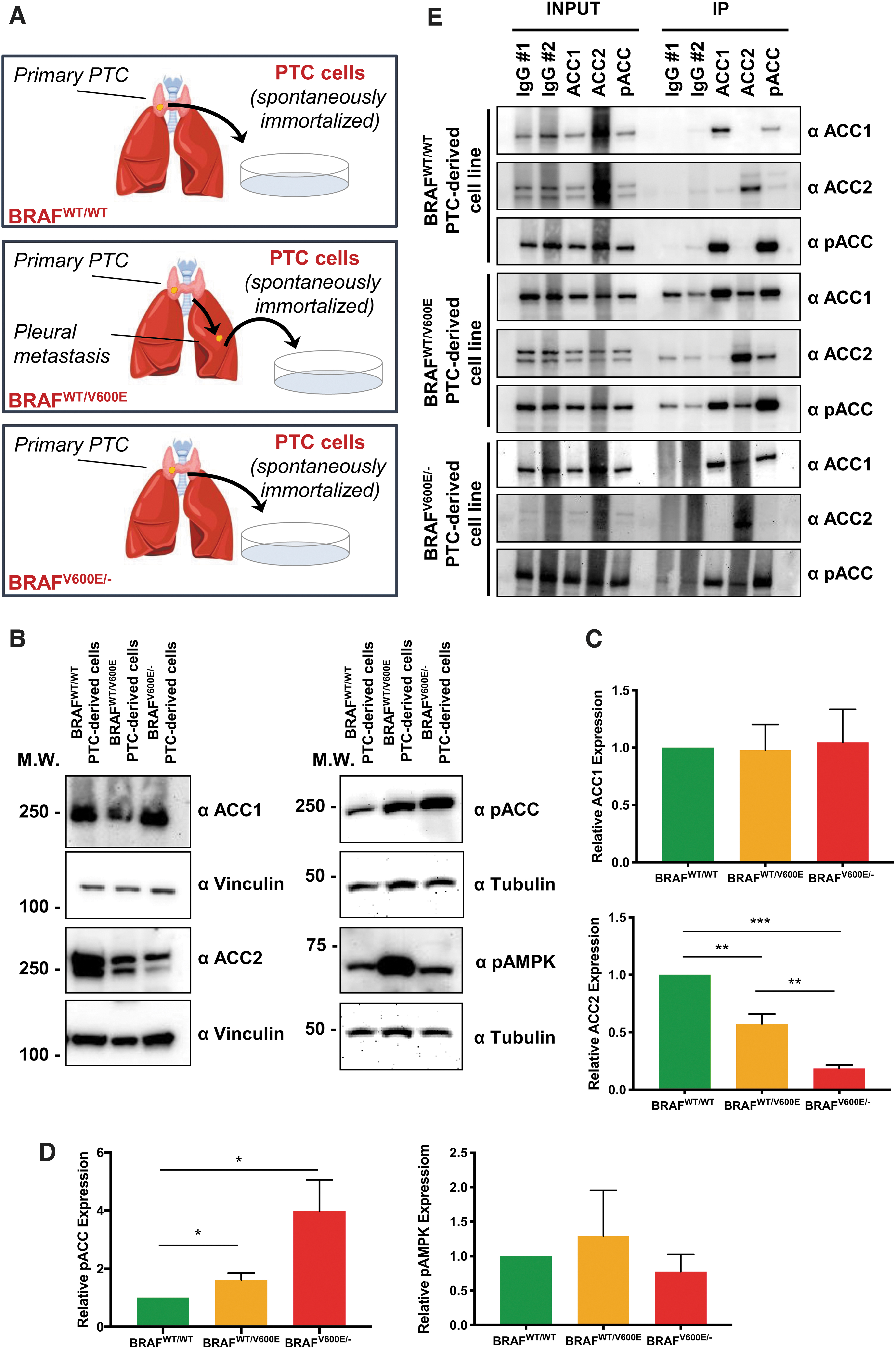
Characterization of ACC1 and ACC2 expression in PTC-derived cell lines with BRAFWT/WT, heterozygous BRAFWT/V600E, or homozygous BRAFV600E/−. (
Targeting BRAFV600E with vemurafenib rescues ACC transcriptional levels in a BRAFV600E allelic dose-dependent manner
All clinical samples of PTC are heterozygous for BRAFV600E mutation (3). We next asked whether selectively targeting BRAFV600E with vemurafenib might modify ACC1 and ACC2 mRNA expression levels observed at baseline in our PTC-derived cell lines. We first measured mRNA expression levels at two time points after treatment with vemurafenib, 4 and 24 hours. Our previous studies showed robust effects on pERK1/2 levels by using 10 μM of vemurafenib compared with lower doses (18). Our metabolic assays here in the current study were performed within 12 hours of vemurafenib treatment to avoid the expected elicitation of resistance, as after this time point we have found in KTC1 heterozygous BRAFWT/V600E PTC-derived cell line a substantial rebound of pERK1/2 levels at 24 hours in the surviving tumor cells, and a further rise at 48 hours (18). Also, we analyzed in this current study pERK1/2 levels in the BCPAP homozygous BRAFV600E/− PTC-derived cell line at 4, 12, and 24 hours (Supplementary Fig. S1).
Here, we found that both ACC2 (five-fold) and ACC1 (two-fold) mRNAs were significantly upregulated by vemurafenib in the homozygous BRAFV600E/− PTC-derived cell line at 24 hours (Fig. 3A, B), but not in heterozygous BRAFWT/V600E and BRAFWT/WT . The increases in ACC2 mRNA levels were higher than ACC1 mRNA levels at 4 hours (1.39-fold in heterozygous or 1.22-fold in homozygous) and 24 hours (1.8-fold in heterozygous or 2.48-fold in homozygous) in BRAFV600E PTC-derived cell lines (Fig. 3A, B). In addition, we analyzed ACC protein levels on treatment with vemurafenib at an intermediate time point (12 hours) to better characterize ACC modulation (Fig. 3C–F). ACC1 protein levels showed no change after vemurafenib treatment (Fig. 3C, D), suggesting that potential post-transcriptional and/or post-translational mechanisms may be involved in the impairment of protein stability. In contrast, ACC2 protein levels were upregulated in both heterozygous and homozygous BRAFV600E PTC-derived cell lines over time (at 24 and 12 hours), with no significant difference observed between vemurafenib and vehicle, perhaps due to the low concentration of serum in the culture media during treatment (Fig. 3E, F). Moreover, we found that at 12 hours, ACC2 protein expression showed a rising trend of increased levels after treatment with vemurafenib compared with vehicle in the heterozygous BRAFV600E PTC-derived cell line (Fig. 3E), although this was not statistically significant across different independent experimental replicates (Fig. 3F); these findings suggest that the genetic heterogeneity of these tumor cells may impact the rescue of ACC2 levels during the therapeutic targeting of BRAFV600E.
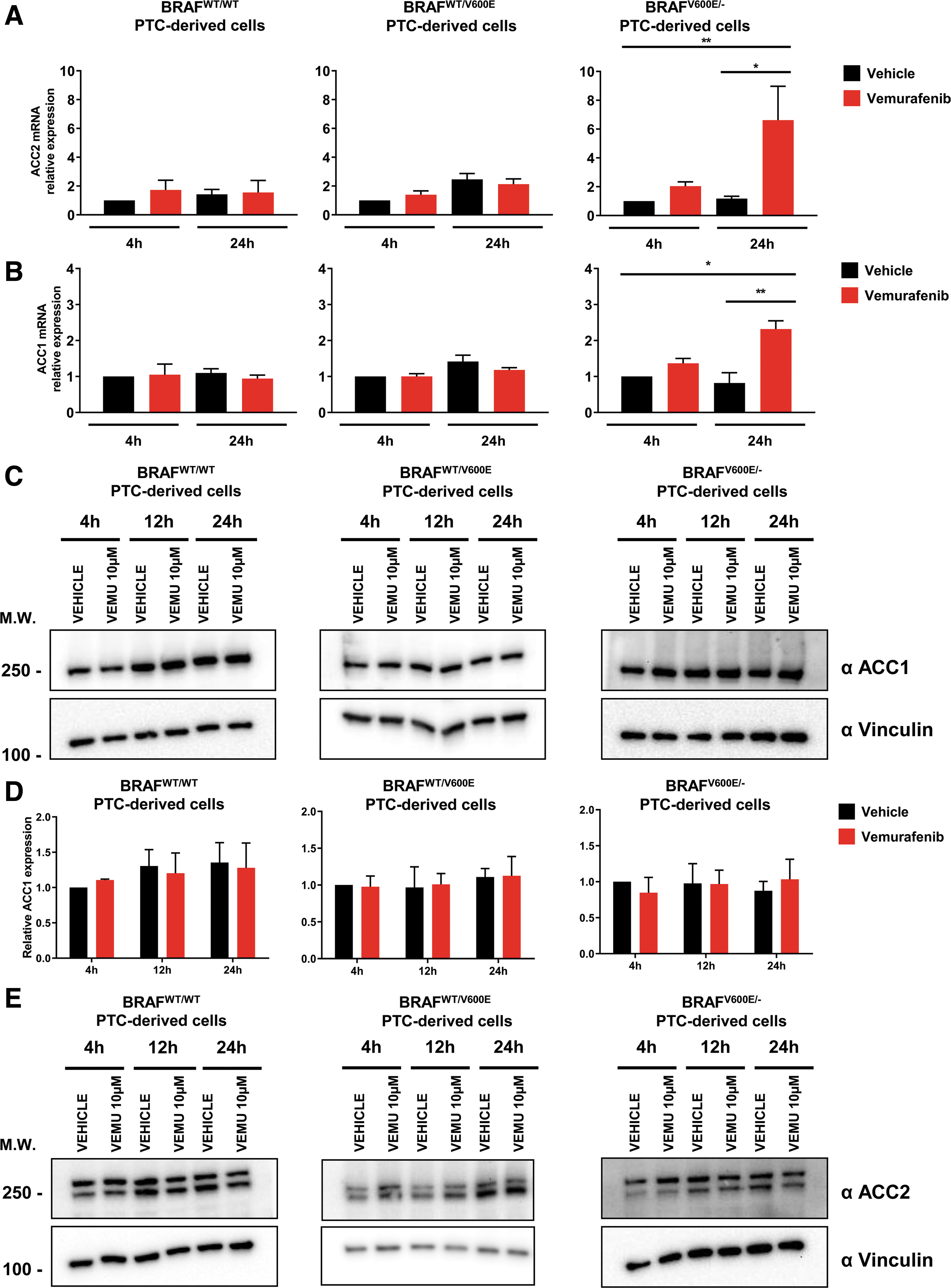
Effects of vemurafenib treatment on ACC1 and ACC2 expression in PTC-derived cell lines. (

To better understand the dynamics of post-translational regulation of both ACC1 and ACC2, we analyzed the expression of pACC and pAMPK (a kinase known to be responsible for ACC phosphorylation and inactivation) and found significantly upregulated phosphorylation of both ACC and AMPK in BRAFV600E PTC-derived cell lines at different time points after vemurafenib treatment (Fig. 3G–H). Overall, these results suggest that ACC2 expression is transcriptionally modulated by vemurafenib in a BRAFV600E allelic-dependent manner, and in addition may be regulated by post-transcriptional and/or post-translational mechanisms.
Targeting BRAFV600E with vemurafenib or ACC gene silencing modulates intracellular ATP and NADPH concentrations, de novo lipid synthesis, FAO, and ROS levels
Since ACC2 transcript levels (and to a lesser extent ACC1) are significantly regulated by BRAFV600E activity in PTC clinical samples, we investigated potential biological phenotypes after treatment with a selective BRAFV600E inhibitor (vemurafenib) or using shRNAs (sh) that efficiently target ACC1 or ACC2 (Fig. 4A–C). First we analyzed ATP intracellular concentration at all time points previously selected (Fig. 4D), since ATP is the molecular unit of currency of intracellular energy transfer and one of the fundamental compounds involved in many metabolic processes, including lipid (fatty acid) synthesis and oxidation. We found that vemurafenib-treated BRAFV600E PTC cells significantly increased ATP concentration at different levels at specific time points (Fig. 4D). This phenotype may be explained by the inhibitory effects of vemurafenib on BRAFV600E, which uses ATP as a donor of phosphate groups to downstream MEK1/2-ERK1/2. Knockdown of either ACC1 or ACC2 did not change ATP levels. Intriguingly, when we measured ACC enzymatic activity, we found that de novo lipid synthesis significantly increased (1.58- and 1.34-fold change in heterozygous and homozygous BRAFV600E -derived cell lines, respectively) within six hours in vemurafenib-treated BRAFV600E PTC-derived cells compared with vehicle (Fig. 4E). The de novo lipid synthesis assay was of short duration (six hours) to avoid major morphological changes in the cell structure by vemurafenib treatment.

Effects of both vemurafenib treatment and knockdown of ACC1 or ACC2 on ATP levels, de novo lipid synthesis, β-oxidation, and intracellular ROS production in PTC-derived cell lines. (
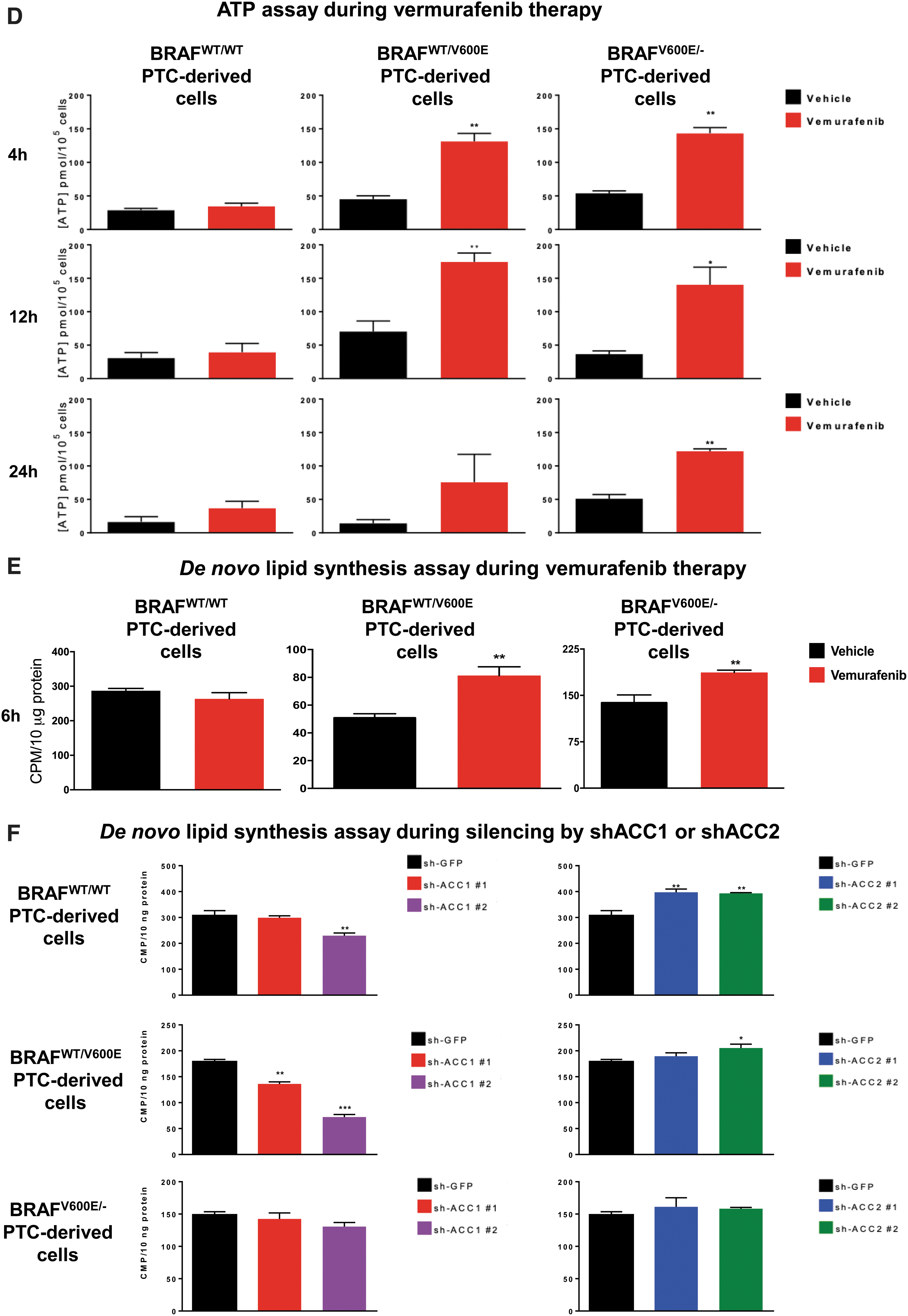



Our shRNA experiments confirmed the specificity of de novo lipid synthesis or FAO (oxygen consumption rate [OCR] and extracellular acidification rate [ECAR]) regulation by ACC1 or ACC2 (Fig. 4F, H, L). As a result of ACC1 or ACC2 molecular action, knockdown (sh) of ACC1 showed significant reductions in de novo lipid synthesis rates mostly in BRAFWT/WT (0.75-fold change, 25% decrease) and heterozygous BRAFWT/V600E (0.76- or 0.4-fold change, 24% and 60%, respectively) cells, which is consistent with the major role played by ACC1 in FAS. Knockdown (sh) of ACC2 instead showed an opposite trend, with significantly increased de novo lipid synthesis rates (1.29-fold change [29%] in BRAFWT/WT and 1.15-fold change [15%] in heterozygous BRAFWT/V600E PTC-derived cells) (Fig. 4F). In contrast, shACC1 or shACC2 did not exhibit any modulation in homozygous BRAFV600E/− PTC-derived cells (Fig. 4F), likely because these cells showed more robust downregulation of basal ACC2 levels (Figs. 2B and 4B) compared with the BRAFWT/WT PTC-derived cell line, and, to a lesser extent, to heterozygous BRAFWT/V600E PTC-derived cells. Results for ACC1 were similar, even if its levels were higher than ACC2 in these cells. In sum, ACC1 or ACC2 knockdown by shRNA had differing impacts in the PTC-derived cell lines; this may be due to additional metabolic genes modulating their regulation of de novo lipid synthesis or β-oxidation. FAO (β-oxidation) was characterized by OCR, and after adding palmitate (PALM):bovine serum albumin (BSA) versus BSA to the assay medium, the BRAFWT/WT PTC-derived cell line showed a preference for oxidative phosphorylation (96% of basal respiration and 93% of maximal respiration) compared with heterozygous BRAFV600E PTC-derived cells. In contrast, 38% of basal and maximum respiration OCR was due to FAO in heterozygous BRAFWT/V600E PTC-derived cells (Fig. 4G). The OCR due to FAO was strongly inhibited in homozygous BRAFV600E/− PTC-derived cells at both basal and maximal respiration (Fig. 4G) compared with heterozygous BRAFWT/V600E PTC-derived cells or BRAFWT/WT PTC-derived cells. Vemurafenib treatment significantly rescued ACC2 mRNA levels compared with vehicle (Fig. 3B) in homozygous BRAFV600E/− PTC-derived cells, although those levels of re-expression were not statistically significant in OCR levels versus vehicle-treated cells (Fig. 4G). Pathways essential for mitochondria respiration may be deregulated in homozygous BRAFV600E/− PTC-derived cells, contributing to the reduction of OCR levels. As a result of this deregulation, these cells may shift to glycolysis. Although OCR due to FAO was higher in the heterozygous BRAFWT/V600E PTC-derived cell line compared with the BRAFWT/WT PTC-derived cell line, critically, their basal respiration OCR due to FAO significantly decreased from 7.95 ± 2.57 pmol/min in the vehicle-treated heterozygous BRAFWT/V600E PTC-derived cells to no utilization of FAO (−0.17 ± 1.26 pmol/min) in vemurafenib-treated heterozygous BRAFWT/V600E PTC-derived cells (p = 0.0154) (Fig. 4G), which also showed a rising trend in ACC2 protein expression levels at 12 hours (Fig. 3E). Similarly, maximal respiration due to exogenous palmitate addition fell significantly, from 7.69 ± 1.92 pmol/min in the vehicle group to 2.64 ± 1.04 in the vemurafenib-treated group (66% decrease, p = 0.0296) (Fig. 4G).
In addition, we found that with silencing (sh) of ACC2 levels both basal and maximum respiration OCR due to FAO were significantly increased (51%, p = 0.0306; 70% p = 0.0341, respectively) in the heterozygous BRAFWT/V600E PTC-derived cell line, while we observed a positive trend in the homozygous BRAFV600E/− PTC-derived cell line (Fig. 4H).
Importantly, mitochondria are both a source and target for oxidative stress. Mitochondrial cellular function can be defined by using a stress test in which the addition of stressors (e.g., vemurafenib, or gene silencing by shRNA, e.g., shACC) can cause alterations in a cell's OCR and bioenergetic profile. With Seahorse technology, we have assessed the levels of FAO through OCR rates (Fig. 4G, H), and glycolysis levels through ECAR rates, as indicators of oxidative stress (measurements of stressed OCR over baseline OCR, and stressed ECAR over baseline ECAR) (Fig. 4I–L). These measurements were performed in vemurafenib-treated PTC-derived cell lines versus vehicle-treated cell lines, or shACC1- or shACC2-transduced PTC-derived cell lines versus shGFP control cell lines with or without addition of exogenous palmitate to the assay medium (Palm:BSA vs. BSA, Fig. 4G–L). In addition to OCR as a metric of energy metabolism, we have also analyzed ECAR (indicator of glycolysis levels after addition of palmitate) rates in three cell lines with different BRAF genetic status. Baseline glycolysis was significantly higher (p < 0.001) in homozygous BRAFV600E/− PTC-derived cells (44.82 ± 3.25 mpH/min) compared with either heterozygous BRAFWT/V600E PTC-derived (14.12 ± 1.40 mpH/min) or BRAFWT/WT PTC-derived cell line (24.87 ± 3.41 mpH/min) (Fig. 4I). Similar results were found in stressed glycolysis (ECAR levels before addition of palmitate), indicating that homozygous BRAFV600E/− PTC-derived cells rely on glycolysis for energy demand. Vemurafenib-treated homozygous BRAFV600E/− PTC-derived cells showed a declining trend in glycolysis (ECAR level), although not at a statistically significant level (Fig. 4I). After utilization of exogenous palmitate, heterozygous BRAFWT/V600E PTC-derived cells showed no significant change in baseline glycolysis (ECAR levels = 14.12 ± 1.40 mpH/min, vehicle Palmitate:BSA) as compared with vehicle BSA (ECAR = 14.58 ± 2.29 mpH/min); while vemurafenib-treated cells showed a significant decrease in glycolysis (ECAR level = −6.27 ± 1.07 mpH/min, p = 0.0085) (Fig. 4I). Similarly, maximal glycolysis due to exogenous palmitate addition was significantly (p = 0.0002) decreased in the vemurafenib-treated heterozygous BRAFWT/V600E PTC-derived cell line (−2.39 ± 0.46 mpH/min) compared with vehicle-treated cells (3.00 ± 0.87 mpH/min), indicating that vemurafenib therapy inhibits glycolysis (Fig. 4I). No changes in ECAR rates were observed in the BRAFWT/WT PTC-derived cell line with vemurafenib treatment (Fig. 4I). In addition, in the heterozygous BRAFWT/V600E PTC-derived cell line with silencing (sh) of ACC2, we observed that after addition of exogenous palmitate, ECAR significantly decreased in both basal (98%, p = 0.031) and maximal glycolysis (71%, p = 0.0177) as compared with shGFP control cells (Fig. 4J), suggesting that ACC2 plays an important role in these metabolic processes.
Collectively, our metabolic data indicate that the heterozygous BRAFWT/V600E PTC-derived cell line uses palmitate for FAO to provide energy (at rates at least two-fold higher than BRAFWT/WT PTC-derived cell line), whereas the homozygous BRAFV600E/− PTC-derived cell line lacks this ability. The homozygous BRAFV600E/− PTC-derived cell line and heterozygous BRAFWT/V600E PTC-derived cell line exhibited lower glucose oxidative phosphorylation levels compared with the BRAFWT/WT PTC-derived cell line. The homozygous BRAFV600E/− PTC-derived cell line showed higher levels of glycolysis compared with both the heterozygous BRAFWT/V600E PTC-derived cell line and the BRAFWT/WT PTC-derived cell line.
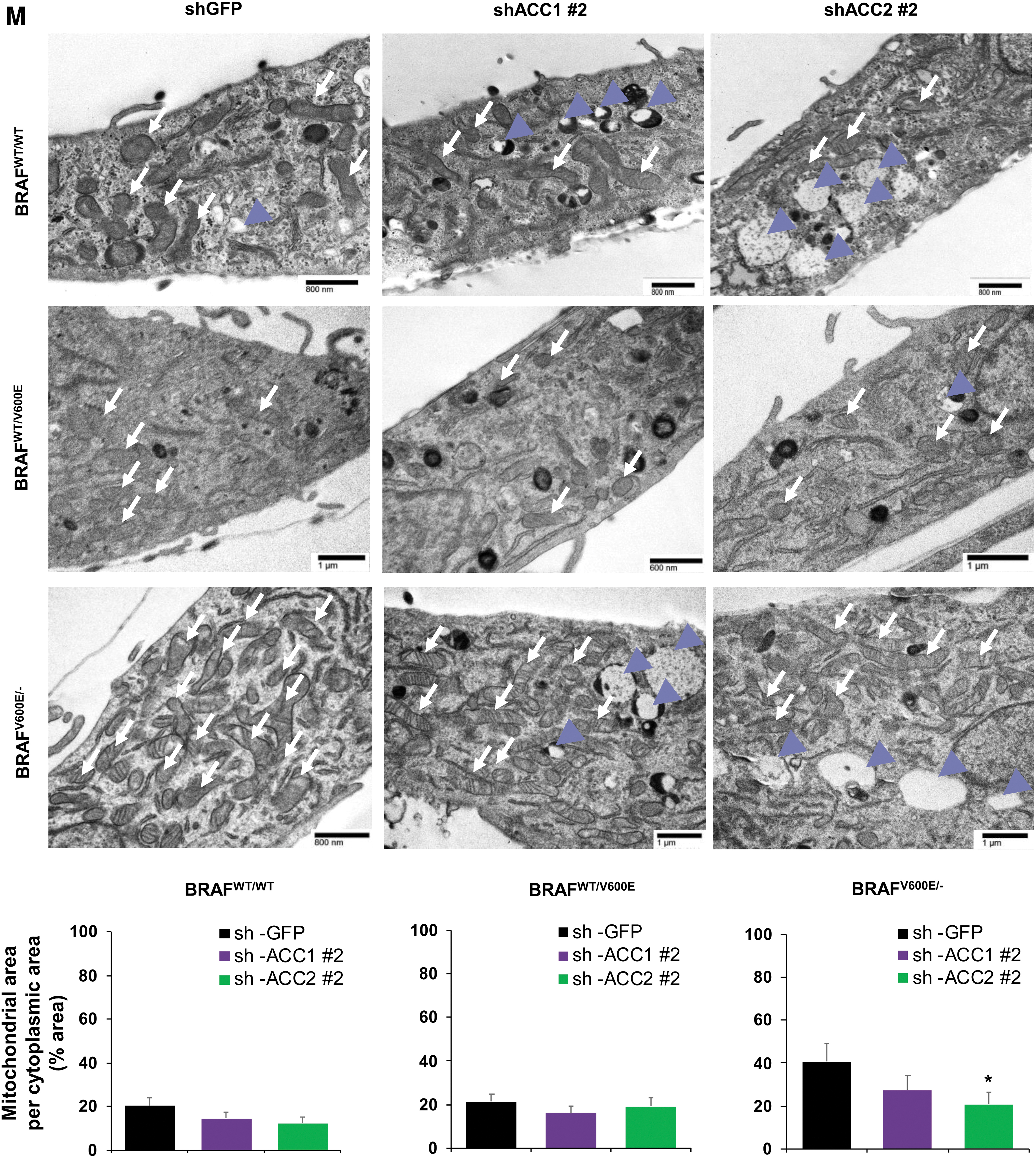
Further, deregulations in mitochondrial respiration machinery are known to influence the intracellular rate of ROS in a membrane potential-independent mode. Therefore, we subsequently investigated a possible redox imbalance in the cells, given the role of ACC1 and ACC2 in consuming and reducing co-factors, which play a fundamental role in maintaining ROS in the cytosol. We further investigated a possible redox imbalance in the PTC-derived cell lines, given the importance of ACC1 and ACC2 in the consumption of NADPH (25). Vemurafenib treatment had no effect on NADP/NADPH ratios in the PTC-derived cells (Supplememtary Fig. S2A), while shACC1 or shACC2 significantly increased NADPH concentrations and decreased NADP/NADPH ratios in the heterozygous BRAFWT/V600E PTC-derived cell line (Supplementary Fig. S2B). To determine whether the fine-tuning of lipid metabolism via ACC enzymes and BRAFV600E affects the oxidative stress of PTC cell lines, we assessed intracellular ROS levels in these cells by using live cell staining assays (Fig. 4K, L). We found that ROS fluorescence intensity was significantly higher (1.48- and 2.15-fold change in heterozygous and homozygous BRAFV600E PTC-derived cell lines, respectively) in vemurafenib-treated BRAFV600E PTC cell lines engineered to express shACC2 versus vehicle-treated shGFP cells (control), suggesting that knockdown of ACC2 may accelerate oxidative stress in BRAFV600E -PTC cells (Fig. 4K, L). Compared with the BRAFWT/WT PTC-derived cell line, BRAFV600E PTC-derived cell lines showed a significant increase in the mitochondrial area in a BRAFV600E allelic-dose dependent manner (i.e., homozygous BRAFV600E/− tumor cells, Fig. 4M), and the knockdown of ACC2 significantly deregulated the ratio between the mitochondrial and cytoplasmic area, and also led to substantial increase in lysosomes (Fig. 4M). There is evidence in the literature that mitochondria and lysosomes can elicit functional cross-talk and converge on mitophagy. How ACC2 impacts this regulation is unknown and will be further investigated in future studies.
ACC2 knockdown contributes to resistance to vemurafenib therapy in a xenograft mouse model derived from a heterozygous BRAFWT/V600E PTC cell line, leading to increased tumor cell growth
Since deregulation of oxidative stress thresholds could contribute to abnormal tumor growth (56), we performed selective knockdowns of ACC1 or ACC2 (Fig. 4A–C) and assessed cell proliferation (Fig. 5A, B). Cell proliferation significantly increased in shACC2 BRAFV600E PTC-derived cell lines treated with vemurafenib (i.e., 1.75 [shACC2 #1] and 1.45 [shACC2 #2] fold changes in the heterozygous BRAFWT/V600E cells, and 3.46 [shACC2 #1] and 2.8 [shACC2 #2] fold changes in the homozygous BRAFV600E/− cells) compared with shGFP vemurafenib-treated cells, with no significant change in BRAFWT/WT PTC cells (Fig. 5A, B). Results from ACC1 knockdown were not as robust across all cell lines (Fig. 5A, B). These results suggest that reduction of ACC2 levels through the BRAFV600E pathway may reduce the inhibitory effect of vemurafenib against thyroid tumor cell viability, and may lead to drug resistance. Further, we selectively overexpressed either ACC1 or ACC2 and found that exogenous expression of ACC2 potently impairs PTC-derived cell lines proliferation, leading to cell growth arrest at a rate of nearly 100% (Fig. 5C, D) whereas the same cell lines engineered with ACC1 overexpression showed a similar proliferation rate to empty vector (control cells).
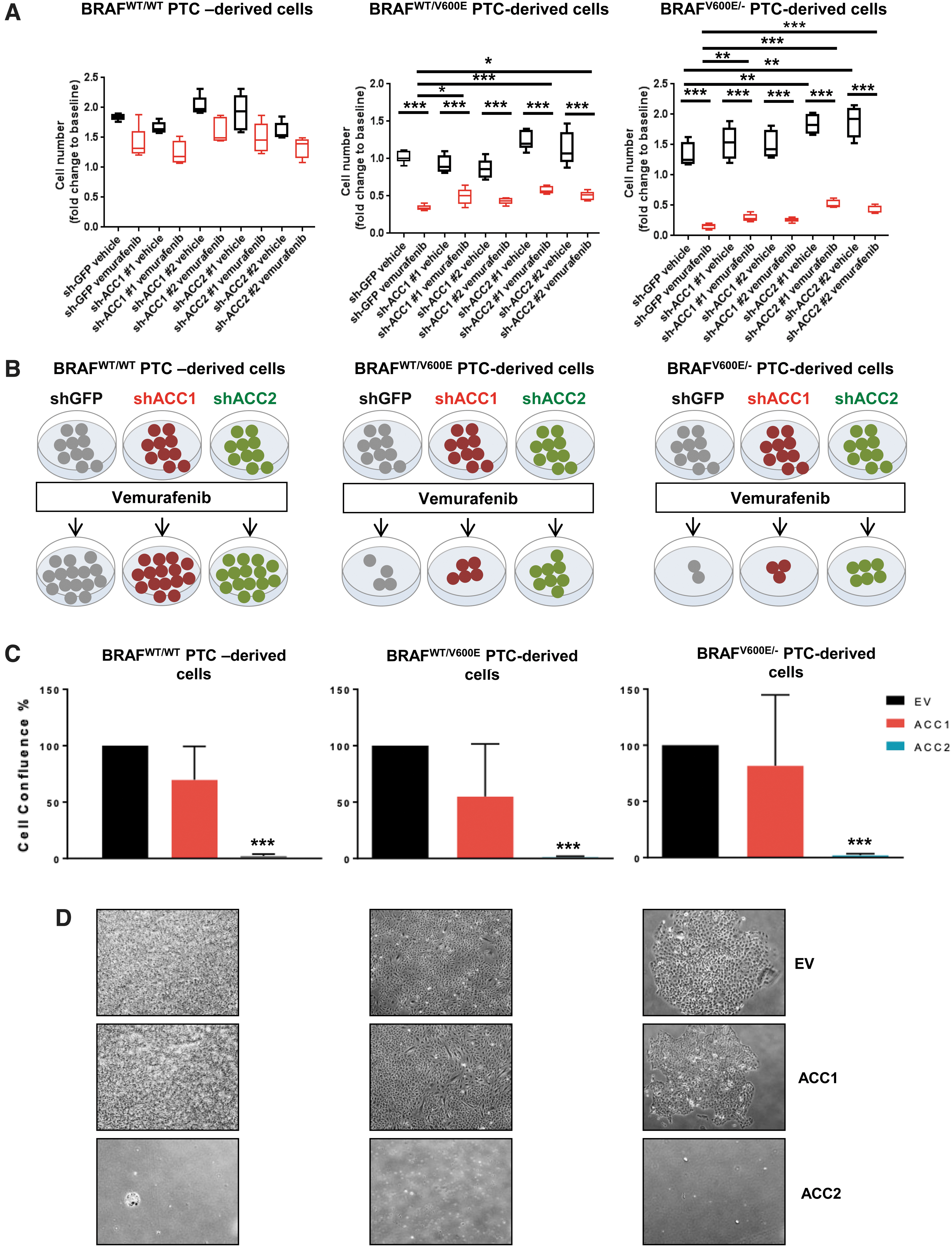
Effects of knockdown or overexpression of ACC1 or ACC2 on tumor cell proliferation in PTC-derived cell lines. (

To assess the impact of ACC1 and ACC2 on tumor growth, and their effects on vemurafenib therapy, we have established the first xenograft mouse model by using the heterozygous BRAFWT/V600E human PTC-derived cell line (i.e., KTC1) engineered with knockdown of either ACC1 or ACC2 (Fig. 5E–G). After two weeks of vemurafenib treatment (Fig. 5E–G), we found no statistical differences in tumor size between the groups of mice implanted with shGFP, shACC1, or shACC2 BRAFWT/V600E human KTC1 cells. Intriguingly, only shACC2 BRAFWT/V600E KTC1 tumors treated with vemurafenib showed increased growth and higher volume (1.44-fold change, p < 0.05) compared with pretreatment baseline, in contrast to the trend observed in vemurafenib-treated shGFP tumors (Fig. 5E–G). shGFP (0.64-fold change) or shACC1 (0.83-fold change) BRAFWT/V600E KTC1 tumors treated with vemurafenib showed decreased tumor volume compared with their baseline. All vemurafenib-treated tumors showed solid nodular components independent of their ACC1 or ACC2 knockdown. Color Doppler sonography displayed blood flows in the peritumoral area within two weeks of treatment in shGFP, shACC1, and shACC2 tumors (Fig. 5G) and showed no statistically significant differences between these groups.
Overall, our findings suggest that downregulation of ACC2, through BRAFV600E, sustains thyroid tumor cell growth (Fig. 6).
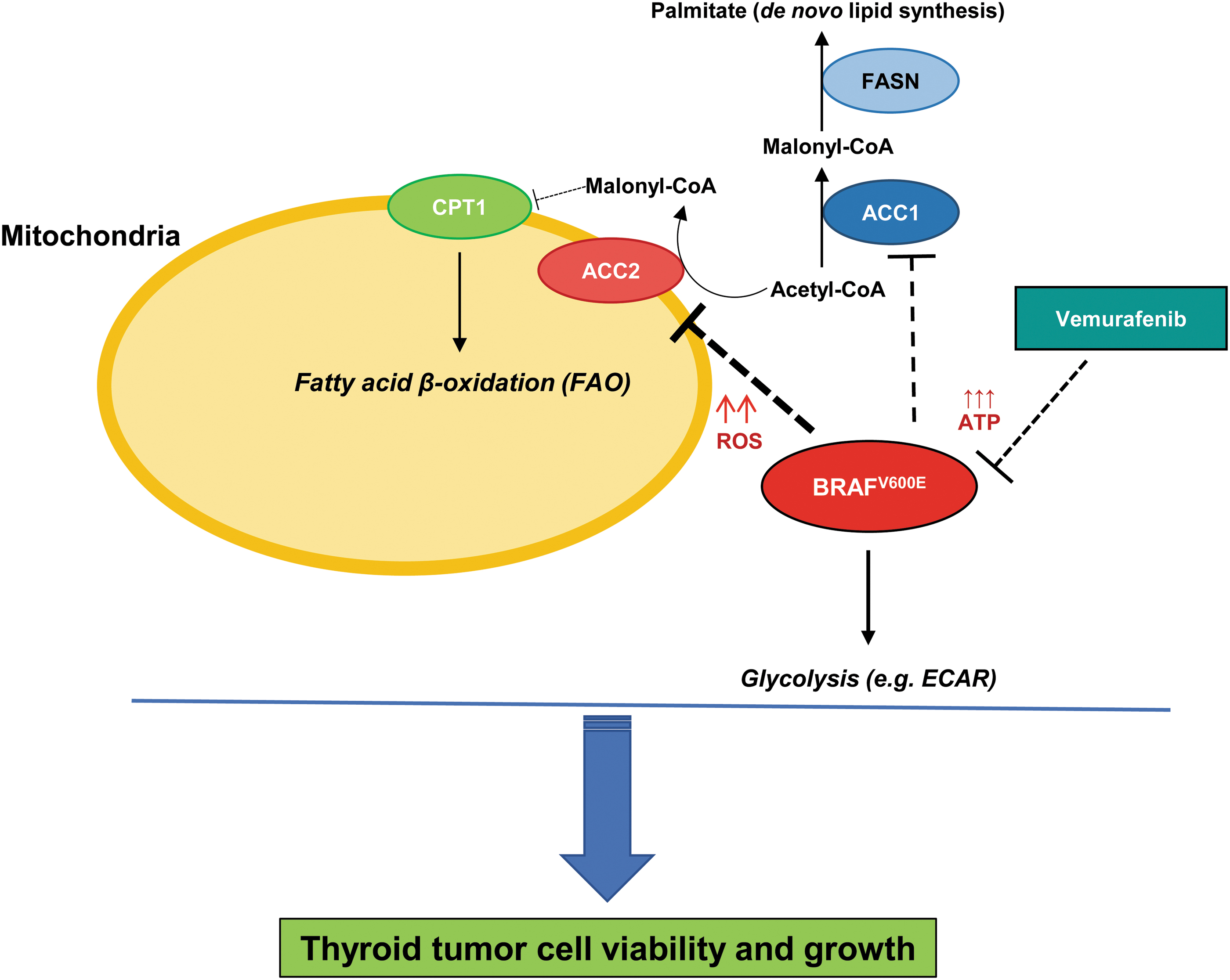
Experimental metabolic model of ACC genes' functions for thyroid carcinoma cell growth. Schematic representation of molecular model of functions between BRAFV600E, ACC1, and ACC2 in human thyroid tumor cell metabolism. Arrows represent modulation, dashed lines represent inhibition, and upward arrows represent increased levels (i.e., ROS and ATP). CPT1, carnitine palmitoyltransferase I; FASN, fatty acid synthase. Color images are available online.
Discussion
Since the initial observations of Otto Warburg (57) describing the aerobic glycolysis phenotype in tumor cells, the characterization of cancer cell metabolism has made huge progress with increasing research attention in recent years (21). BRAF is an ATP-dependent cytosolic kinase that regulates the MAPK (MEK1/2 and ERK1/2) signaling pathway; the BRAFV600E-MEK-ERK pathway is characterized by the loss of negative feedback inhibition and is potently causative of PTC cell dedifferentiation (58). Fortunately, BRAFV600E-selective inhibitors (e.g., vemurafenib) are clinically available for treatment of BRAFV600E -positive tumor patients; however, drug resistance mechanisms are still an important issue in oncology (6,9,10,19,59). Balance and inter-functions between de novo lipid synthesis (e.g., FAS) and FAO, and their connection to prime genetic events such as the BRAFV600E mutation, still need to be deeply investigated. Lipids are an important energy resource for cells: They are crucial for membrane biogenesis, and their oxidative catabolism provides ATP and reducing equivalents in the form of NADH, both of which are essential to controlling environmental stress and promoting survival (25,28). ACC2 expression was downregulated in BRAFV600E -PTC clinical samples and in BRAFV600E -PTC-derived cell lines compared with BRAFWT -PTC, whereas ACC1 levels were not. We also found an upregulation of ACC2 mRNA levels (and to a lesser extent ACC1) in a BRAFV600E allelic-dosage-dependent manner in PTC-derived cells when treated with vermurafenib. So it is likely that ACC genes' expression is influenced by the BRAFV600E pathway. Further studies will be needed to test this mechanism-based hypothesis and the role of ACC2 in primary resistance exhibited by BRAFV600E -PTC cells. The IP of ACC2 indicated that its phosphorylation may occur only in BRAFV600E -PTC-derived cells, suggesting that BRAFV600E may also orchestrate post-translational regulations of ACC2 (and ACC1, in part). Intriguingly, levels of phosphorylated (p) ACC (known to be inactive) increased in both heterozygous and homozygous BRAFV600E tumor cells when we used vemurafenib. However, the majority of ACC2 levels may be nonphosphorylated in BRAFV600E -PTC cell lines, as suggested by our IP studies, which showed that most of the phosphorylation targeted the ACC1 protein. Therefore, it is likely that ACC2 is functionally more active than inactive (because less phosphorylated), and it contributes to downregulation of FAO and upregulation of de novo lipid synthesis. This may lead to decreased tumor cell survival. Phosphorylated ACC showed a positive trend with levels of phosphorylated AMPK, which is a crucial energy sensor that works as a metabolic checkpoint for most eukaryotic cells (60). A critical metabolic function of all cells is balancing ATP consumption and ATP production. AMPK is a highly conserved sensor of intracellular adenosine nucleotide levels that is activated when reductions in ATP production determine relative increases in AMP or ADP. One major mechanism of AMPK includes phosphorylation of ACC1 and ACC2 to inhibit FAS and promote FAO (61). Moreover, AMPK can be activated by direct phosphorylation (Thr-172 of AMPKalpha) from the tumor suppressor LKB1 serine/threonine kinase (62). Interestingly, the AMPK pathway and its target pACC are upregulated in PTC compared with NT samples (63). Early in vemurafenib treatment, BRAFV600E PTC-derived cell lines reversed their metabolic phenotype: de novo lipogenesis increased and FAO due to OCR decreased along with ECAR rates. This metabolic mechanism may counterbalance potential over-activation of oxidative stress (i.e., ROS levels), which may lead to cytotoxicity toward BRAFV600E -thyroid tumor cells. Critically, knockdown of ACC2 levels led to significantly increased BRAFV600E tumor cell proliferation and ROS production during vemurafenib therapy, indicating that silencing of this gene may enhance tumor survival and proliferation. Importantly, our xenograft mouse data further showed that human BRAFV600E -thyroid tumor cells became less responsive to vemurafenib within two weeks, and they ultimately exhibited increased tumor growth when the ACC2 gene was knocked down. Meanwhile, overexpression of ACC2 (but not ACC1) led to strong inhibition of proliferation in PTC-derived cell lines. The role of ROS in cell biology emerges as a double-edged sword, as ROS can trigger tumor cell proliferation or death. There is evidence that cellular levels of ROS are of critical importance in thyroid carcinoma (64). What matters most regarding ROS depends on the threshold required for its maintenance in tumor cells.
Collectively, our work has demonstrated for the first time that the downregulation of ACC2 levels via BRAFV600E plays a critical role in PTC-derived cells, and establishes favorable conditions for thyroid tumor cell proliferation. These findings suggest a potential link between BRAFV600E and lipid metabolism regulation in PTC. Silencing of ACC2 may contribute to BRAFV600E inhibitor (e.g., vemurafenib) resistance and increased tumor growth. ACC2 rescue may represent a novel molecular strategy for overcoming resistance to BRAFV600E inhibitors.
Footnotes
Acknowledgments
The authors thank Rork Kuick (University of Michigan) for downloading and organizing (see details in Materials and Methods) ACACA (ACC1) and ACACB (ACC2) TCGA copy number and RNA-seq data. Veronica Valvo was recipient of a PhD fellowship from the MIUR and UCSC (Roma, Italy). The authors thank Stephanie Li and Elizabeth McGonagle (undergraduate students) for technical assistance. They thank Maria Ericsson (Harvard Medical School, Boston) for the cell block preparation for the transmission electron microscopy.
Authors' Contributions
Conception and design: C.N.; Writing the article: V.V. and C.N.; Editing the article: all authors; Revision of the article: all authors; Development of methodology: V.V., A.I., T.R.K., C.P., Z.Z., X.L., and C.N.; Acquisition of data: V.V., A.I., T.R.K., C.P., Z.Z., I.E.S., S.D.B., X.L., and C.N.; Analysis and interpretation of data: all authors.
Author Disclosure Statement
Carmelo Nucera was NIH ad hoc peer reviewer. No potential conflicts of interest were disclosed by the other authors.
Funding Information
Carmelo Nucera (Principal Investigator, Human Thyroid Cancers Preclinical and Translational Research at the Beth Israel Deaconess Medical Center [BIDMC/Harvard Medical School]) was awarded grants by the National Cancer Institute/National Institutes of Health (1R01CA181183-01A1 and R01CA248031-01), the American Thyroid Association (ATA), and ThyCa:Thyroid Cancer Survivors Association Inc., for Thyroid Cancer Research. Carmelo Nucera was also a recipient of the BIDMC/CAO Grant (Boston, MA).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2