
Abstract
Background:
Antigen-specific lymphocytes are increasingly investigated in autoimmune diseases and immune therapies. We sought to identify thyrotropin receptor (TSHR)-specific lymphocytes in mouse models of Graves' disease, including Graves' patient-specific immunotype human leukocyte antigen (HLA)-DR3, and in frozen and thawed Graves' patient blood samples.
Methods and Results:
Splenic lymphocytes of adenovirus (Ad)-TSHR-immunized BALB/c mice were stimulated with TSHR-specific peptides C, D, or J. Furthermore, CD154-expressing cells were enriched, expanded in vitro, and analyzed for binding of peptide-major histocompatibility complex (MHC) II multimers (“tetramers,” immunotype H2-IAd). Only peptides C and J were able to elicit increased expression/secretion of CD154 and interferon-γ, and tetramers which were loaded with peptide C resulted in antigen-specific signals in splenic lymphocytes from Ad-TSHR-immunized mice. Accordingly, TSHR-specific HLA-DR3-MHC class II tetramers loaded with peptide p10 specifically bound to human HLA-DR3-(allele B1*03:01)-transgenic Bl/6 mouse splenic T lymphocytes. In addition, we fine-tuned a protocol to reliably measure thawed human peripheral blood mononuclear cells (PBMCs), which resulted in reliable recovery after freezing and thawing with regard to vitality and B and T cell subpopulation markers including regulatory T cells (CD3, CD4, CD25, FoxP3, CD25high, CD127low). TSHR-specific HLA-DR3-MHC class II tetramers loaded with peptide p10 identified antigen-specific T cells in HLA-DR3-positive Graves' patients' thawed PBMCs. Moreover, stimulation-dependent release of interleukin (IL)-1beta, IL-6, tumor necrosis factor-alpha from thawed PBMCs occurred at the expected levels.
Conclusions:
Novel MHC II tetramers identified TSHR-specific T lymphocytes in Ad-TSHR-immunized hyperthyroid BALB/c or HLA-DR3-transgenic mice and in thawed human PBMCs from patients with Graves' disease. These assays may contribute to measure both disease severity and effects of novel immune therapies in future animal studies and clinical investigations of Graves' disease.
Introduction
Antigen-specific lymphocytes are increasingly investigated in autoimmune diseases and immune therapies. We sought to identify thyrotropin receptor (TSHR)-specific lymphocytes in animal models of Graves' disease and in blood samples of patients with Graves' disease.
Mouse models of Graves' disease have been established through immunization with DNA encoding the human TSHR or its A-subunit encoded by plasmid or adenoviral vectors (1,2). Following up on extensive characterizations of two or three times adenovirus (Ad)-TSHR-immunized mice (1,2), our group has established a long-term model of Graves' disease and orbitopathy (3,4). So far, several studies have described measurements of B and T lymphocytes and their subsets, for example, regulatory T cells, in animal models of Graves' disease (2,5,6) and in human blood samples of patients with Graves' disease (7,8). However, these investigations have reported general regulation of cell types, but not of antigen-specific cells, with the exception of one study which used major histocompatibility complex (MHC) class I-specific tetramers (9).
Because MHC II epitopes might be more relevant for autoimmunity, and during tolerance induction, we wished to test the feasibility of detecting antigen-specific immune cells in either normal BALB/c mice or human leukocyte antigen (HLA)-DR3-transgenic mice using peptide-MHC class II multimers (further named tetramers). MHC class II proteins are considered to be critically involved in mechanisms of autoimmunity, and possibly induction of tolerance. Previous work had described antigen-specific TSHR-derived epitopes (10,11) in the established BALB/c mouse model, which possesses the immunotype H2-IAd. We first verified that the same epitopes were identified in our long-term immunization mouse model and then established a protocol of cell isolation, enrichment, and in vitro expansion to identify antigen-specific immune cells using MHC class II tetramers. In this procedure, analysis and expansion of T lymphocytes were partially based on methods described in previous reports (12,13).
Moreover, we also wished to investigate the usability of frozen and thawed human peripheral mononuclear blood cells (PBMCs) to study B and T lymphocyte populations, as well as regulatory T cells (Treg cells). All populations were analyzed in freshly isolated cells and in cells after freezing, storage at −80°C, and thawing, by using viability staining and flow cytometric measurements. So far, several studies have investigated the feasibility of using thawed PBMCs, and several protocols are available (e.g., 14–16). For example, Hønge et al. (17) found that small changes of the thawing procedure significantly impact PBMCs for specific fluorescence-activated cell sorting (FACS) measurements. Since the variations between different protocols may impact on the quality of measurements of (antigen-)specific lymphocyte subpopulations, we established a single method to investigate the proportion of B and T cells including CD4+CD25highCD127lowFoxP3+ Treg cells in thawed human PBMCs after storage at −80°C. This protocol was chosen from a number of alternative method descriptions, and this article presents the resulting optimized method. One aspect was to include a SepMate® ready-to-use kit, which should improve feasibility and reproducibility of future blood sampling at various clinical centers compared with previous protocols (14,15).
Using established TSHR-specific peptide epitopes in HLA-DR3-transgenic mice (18,19), we also conceived novel MHC class II DR3 (DRB1*03:01) tetramers to measure specific T cells in these mice. This assay was then used to measure antigen-specific cells in human blood samples of HLA-DR3-expressing patients with Graves' disease.
Generally, the aim was to establish a protocol which would allow for easy and standardized analysis of spleen cells from mouse Graves' disease models and of heparin- or EDTA-treated blood sampled from patients with Graves' disease of future multicenter clinical trials of antigen-specific interventions.
Materials and Methods
Mouse cells: antibodies and peptides
Please see the Supplementary Data, and mouse antibodies, as shown in Table 1.
Mouse Lymphocyte Antibody Markers
APC, allophycocyanin; FITC, fluorescein isothiocyanate; IFN, interferon; PE, phycoerythrin; PerCP, peridinin-chlorophyll-protein.
HLA-DR3-transgenic mice
HLA-DRB1*0301-transgenic mice (further named HLA-DR3 mice) on a C57Bl/6 background were kindly provided by Dr. Grunwald, Fraunhofer Institute Leipzig. This mouse strain is based on the one described in previous publications (20,21). Animals were kept under standard housing conditions (23°C ± 2°C, 55% ± 10% relative humidity) in groups of 10 animals in GR1800DD cages (Tecniplast®).
All animal experiments were approved by the local animal welfare authority and ethics committee of the Regierung von Oberbayern (Government of Upper Bavaria) in Munich, Germany (no. 55.2-1-54-2531-25-12), and carried out in accordance to the World Medical Association (Declaration of Helsinki) and the European Commission guidelines (Directive 2010/63/EU). All guidelines for care of animals were respected.
Induction of symptoms of hyperthyroidism with THSR-expressing adenovirus, isolation of mouse spleen cells, and stimulation of spleen cells for intracellular detection of activation markers: please see the Supplementary Data.
Intracellular detection of activation markers CD154 and IFN-γ, CD154 enrichment and expansion of activated cells, and viability staining of CD154-enriched and expanded cells: please see the Supplementary Data.
Peptide-MHC II multimers (further named tetramers)
MHC class II immunotype H2-IAd tetramers were conceived on the basis of previous stimulation experiments using peptides C, D, or J. This immunotype is present in BALB/c mice. allophycocyanin (APC)-labeled H2-IAd/pep C, H2-IAd/pep D, H2-IAd/pep J, and H2-IAd/CLIP87-101 tetramers were obtained through the National Institute of Health core facility at Emory University, Druid Hills, Atlanta, GA.
For MHC staining, wells containing CD154-enriched, expanded, and stained murine splenic lymphocytes were washed once with phosphate-buffered saline (PBS), 5% fetal calf serum (FCS). Cell pellets were suspended in 45 μL PBS, 5% FCS. Per well, 5 μL pMHCII-APC tetramers—H2-IAd/pep C, H2-IAd/pep D, H2-IAd/pep J (1:10 in PBS/5% FCS; final 14 μg/mL), or Clip control tetramer (15 μg/mL final) were added and the plate was incubated for 40 minutes at 2–8°C. Ten microliters of anti-CD4-antibody phycoerythrin (PE) (1:100) and anti-CD3 antibody-fluorescein isothiocyanate (FITC) (1:20) in PBS, 5% FCS, were added and samples were incubated for another 20 minutes at 2–8°C. Wells were washed by the addition of 200 μL PBS/5% FCS and a second time with 250 μL. Cell pellets were suspended in 200 μL PBS/1% bovine serum albumin/1% PFA and analyzed by flow cytometry after compensation.
Determination of T4 values and anti-TSHR antibody titers from serum of mice
Please see the Supplementary Data.
Phenotyping of HLA-DR3 mice
Please see the Supplementary Data.
Genotyping of HLA-DR3 mice
Genotyping of HLA-DR3-transgenic mice was performed by polymerase chain reaction (PCR) using the primer pair DRB1*03-s and DRB1*03-as for HLA-DR3, as shown in Table 2. Control reactions with primers oIMR7338 and oIMR7339 (derived from the sequence of mouse interleukin [IL]-2) were performed in separate tubes to see whether the DNA allowed for amplification. HLA-DR3-specific primers were synthesized at Eurofins Genomics, and control primers were purchased from Invitrogen.
Polymerase Chain Reaction Primer Pairs Used for Genotyping of DR3 Mice
Control primers oIMR7338 and oIMR7339 were derived from the sequence of mouse interleukin-2.
To obtain DNA of each mouse, freshly taken ear cuts were suspended in 100 μL Snooplex FastPrep lysis buffer (GVG Genetic Monitoring) and incubated at 80°C for 20 minutes. 0.6–3 μL of the lysed samples were used as PCR templates. PCRs were performed in 1 × Herculase reaction buffer with 200 μM dNTP, 20 pmol of each primer, and 0.5 μL Herculase II Fusion Enzyme (no. 600677; Agilent Technologies). PCR products were subsequently analyzed on a 2% Agarose High Resolution gel (no. K297; Carl Roth).
Depletion of CD8-positive cells and staining with peptide MHC II multimers (tetramers)
To use magnetic cell separation (MACS) MS columns, splenic lymphocytes of each HLA-DR3-transgenic mouse were divided in five aliquots of 1 × 107 cells each and suspended in 90 μL MACS buffer. Ten microliters of anti-CD8 MicroBeads (no. 130-117-044; Miltenyi) were added and samples were incubated at 4–8°C for 15 minutes. Cells were washed with 1.5 mL MACS buffer, centrifuged, and suspended in 500 μL MACS buffer. The suspension was added on top of an MS column previously equilibrated with 500 μL MACS buffer. The flow through and the first wash fraction were collected as CD8-depleted/reduced cell fraction.
Cells were sedimented and suspended in 240 μL 50 nM dasatinib in PBS. Aliquots of 4.17 × 106 cells per well (minus missing CD8+ cells) were prepared on V-bottom 96-well plates for incubation with tetramers in duplicates.
The plate was incubated for 30 minutes at 37°C, then 3 μL of pMHCII tetramers (see next paragraph; final 5.9 μg/mL, Clip, p10, p37, pE, or pI) were directly added, and incubation at 37°C was prolonged for 1 hour. Cells were washed by the addition of 150 μL PBS and centrifuged. Cell pellets were suspended in 200 μL FVS780 (1:3000 in PBS) and incubated at room temperature (RT) in the dark for 10 minutes. After centrifugation, wells were washed once with 250 μL MACS buffer. Cells were suspended in 100 μL staining solution (10 μL FcR Blocking, 6 μL anti-CD3/FITC, 3 μL anti-CD4/PerCP in MACS buffer) and incubated at 4°C for 17 minutes. Wells were washed once with 150 μL and a second time with 250 μL MACS buffer. Cells were suspended in 250 μL MACS buffer and analyzed by flow cytometry after compensation of the device.
HLA-DR3 tetramers for human TSHR-derived peptides
APC-labeled ProT2® MHC class II DR3 tetramers (pMHCII-APC) were synthesized at ProImmune Ltd., Oxford, United Kingdom. They were produced with bound peptides 10, 37, E, or I (Table 4B) and were composed of four molecules each of DRA1*01:01 α-chain and DRB1*03:01 β-chain, respectively. One chain was biotinylated and complexes of four molecules formed by streptavidin binding. A tetramer with bound Clip(87-101) peptide was used as negative control tetramer.
Peptide Sequences and Thyrotropin Receptor Locations Used to Construct Major Histocompatibility Complex II Tetramers
TSHR, thyrotropin receptor.
Human lymphocytes: antibodies
Used markers are listed in Table 3. All antibodies were from BD Biosciences except clone VIT4 and TÜK4, which were from Miltenyi Biotec.
Human Lymphocyte Antibody Markers
Additional material
Please see the Supplementary Data.
Graves' patient blood samples
Blood samples (20 mL each) from 3 HLA-DR3-positive patients (2 female, 1 male, aged 40–60 years) with Graves' disease were collected after ethics approval 7022/2016/EKU (0102/16) by the Bavarian Medical Chamber. The principles of the Declaration of Helsinki were respected. Consent has been obtained from each subject after full explanation of the purpose and nature of all procedures used. All patients had clinical signs of hyperthyroidism such as tachycardia; they were nonsmokers. The body mass index was within the reference range. Diagnosis of Graves' disease was confirmed by detection of relevant titers of anti-TSHR antibodies in all patient sera. Samples from healthy volunteers served as controls.
Preparation of PBMCs with SepMate
Please see the Supplementary Data.
Freezing and thawing of cells
For the freezing procedure, solutions, vials, and the cryobox were precooled (4°C). Volumes were adjusted as appropriate according to cell count. Two milliliters of cells (2 × 107 cells/mL) were centrifuged at 350 g for 5 minutes. The cell pellet was resuspended with 1 mL of FCS to obtain a cell count of 4 × 107 cells/mL. One milliliter of FCS with 20% dimethyl sulfoxide (DMSO) was added dropwise to the cells while swirling the tube (final concentration of DMSO was 10%). Cells were frozen in aliquots and put into a cryobox, which was transferred into the −80°C deep freezer. After incubation overnight, the vials were transferred into the liquid nitrogen tank for long-term storage or kept at −80°C for short-term storage.
For thawing of PBMCs, cryovials were removed from the liquid nitrogen tank and transferred into the precooled (−80°C) cryobox. In case of thawing several vials, only one vial was processed at a time. The vials were thawed in a water bath at 37°C. Almost completely thawed cells were transferred immediately into a 15 mL tube and 10 mL of warm (RT) PBS containing 10% heat-inactivated FCS were added fast but dropwise to the tube while swirling. To remove DMSO, cells were washed by centrifuging the tubes for 8 minutes at 300 g. The cell pellet was resuspended in 1 mL MACS buffer containing 2% human serum pool (2 × 107 cells/mL) with or without 0.02% sodium azide.
Viability stain
Please see the Supplementary Data.
Staining of cells with B or T cell markers (fluorescence-labeled antibodies)
Staining of B cell populations was carried out in MACS buffer with 2% human serum pool on a 96-well plate by adding 5 μL anti-CD3-APC, 5 μL anti-CD19-PerCP-Cy5.5, 5 μL anti-CD27-PE-Cy7, and 20 μL anti-IgD-FITC to 106 cells in a volume of 100 μL. T cell populations were stained using 5 μL anti-CD3-APC, 3.7 μL anti-CD4-PerCP-Cy5.5, and 3.7 μL anti-CD8-APC-Cy7 in the same volume. Single-stained controls and fluorescence marker only controls were processed in parallel. Cells were incubated for 15 minutes in the dark at RT or at 4°C. After washing twice by filling the wells ad 250 μL/well with MACS buffer, cell pellets were resuspended in 200 μL MACS buffer and the plate was analyzed by flow cytometry counting at least 100,000 cells in the lymphocyte gate. Gating of B cells was performed as recommended and described by BD Biosciences (B cell research, flow cytometry tools for the study of B cell biology, BD, 2014) and described in Maecker et al. (22).
Staining of Treg cells
Treg cells were identified by the combination of the markers CD4+CD25high CD127lowFoxP3+. In a first step, 1 × 106 FVS780 processed cells were stained with 1 μL CD4-PerCP (prediluted 1:2), 5 μL anti-CD25-BB515, 5 μL anti-CD127-PE-Cy7, or 5 μL isotype control antibodies in a total volume of 100 μL for 20 minutes in the dark at 4–8°C. To identify CD14-expressing cells, 9 μL of anti-CD14-APC-Vio770 were used in addition to cells, which had not been stained with FVS-780 because both dyes are measured in the same fluorescence channel. Cells were washed twice with MACS buffer by filling the wells ad 250 μL/well and centrifuged. In a second step, cells were fixed and permeabilized by the addition of 0.3 mL Fix/Perm Buffer and incubation for 40–50 minutes at 4–8°C in the dark. After washing wells twice with 250 μL Perm/Wash Buffer, cells were stained in a total volume of 100 μL for intracellular FoxP3 by adding 18 μL anti-FoxP3-PE antibody or isotype control antibody and incubation for 40–50 minutes in the dark at 4–8°C. After washing twice as before, cell pellets were resuspended in 200 μL MACS buffer and the plate was analyzed by flow cytometry after a compensation procedure using single stained controls.
Genotyping for HLA-DR3 and tetramer staining
Please see the HLA-DR3-transgenic mice section.
Stimulation of PBMCs and measurement of secreted cytokines
Freshly isolated or thawed PBMCs were suspended in growth medium (VLE RPMI 1640, 10% heat-inactivated FCS, 100 U/mL penicillin, 100 μg/mL streptomycin) at a living cell density of 106/mL. One milliliter was seeded to each well of a 24-well plate. Cells were stimulated by the addition of 1 μg/mL anti-CD3 antibody in combination with 5 μg/mL anti-CD28 antibody, 10 μg/mL Zymosan, or 100 ng/mL lipopolysaccharide (LPS). The plate was incubated at 37°C, 5% CO2 for 18 hours. Supernatants were harvested, centrifuged to remove particulate material, and stored frozen in aliquots at −20°C. Analysis for IL-1beta, IL-6, and tumor necrosis factor (TNF)-alpha of undiluted or diluted supernatants was performed using commercially available enzyme-linked immunosorbent assay (ELISA) kits from BD Biosciences according to the supplier's manual.
Statistics
The analyses of this study including differences in expression levels of markers on freshly isolated and thawed cells were checked using analysis of variance and Student's t-test for paired samples where appropriate.
Results
BALB/c mouse splenic cells
Verification of hyperthyroidism
As described before (3,23), immunization of BALB/c mice with Ad-TSHR, but not Ad-green fluorescent protein (GFP) led to clinical symptoms of hyperthyroidism. Determination of anti-TSHR antibody titers revealed that all analyzed Ad-TSHR-immunized animals were positive on day 56 and 77 after first immunization with varying titers. Ad-GFP-immunized animals were anti-TSHR antibody negative. At day 56 after the first immunization, all except one animal had a thyroxine (T4) value above the cutoff of 7.93 U/L, which was determined on the basis of the mean T4 values observed in Ad-GFP-immunized mice.
Splenic lymphocyte responses to TSHR-specific peptides
Peptides C, D, and J derived from the sequence of the human TSHR had been described as T cell epitopes in BALB/c mice by Pichurin et al. (10)—the sequences are shown in Table 4A. We selected these peptides to confirm specific stimulation of mouse spleen cells for further study of antigen-specific T cells by using H2-IAd tetramers coupled to these epitopes.
Isolated spleen cells of immunized BALB/c mice were stimulated with 1 μg/mL of each peptide. Intracellular levels of interferon

Intracellular expression levels of CD154 and IFN-γ after peptide stimulation. Isolated spleen cells from BALB/c mice, which had been immunized with Ad-TSHR, Ad-GFP, or had not been immunized (“native”), were stimulated for 8 hours with 1 μg/mL of TSHR-specific peptides C, D, or J and subsequently analyzed for intracellular expression of the activation marker CD154 and of the cytokine IFN-γ by FACS. Vehicle alone was used as control. (
CD154 enrichment by in vitro cultivation and peptide-MHC II multimer (tetramer) staining
Pools of splenic cells from immunized mice were stimulated with 1 μg/mL peptide or pools of 1 μg/mL of each peptide (for controls), or PMA/ionomycin in the presence of a PE-labeled monoclonal antibody against the T cell activation marker CD154, which included an anti-CD40 antibody to block interaction with CD154. After overnight incubation, cells were enriched using magnetically labeled anti-PE MicroBeads.
The mean enrichment factor of CD4-positive T cells in unstimulated cells of Ad-TSHR-immunized mice was 11.4-fold, in cells stimulated with peptides C and J, these rates were 24.2 and 16.6, respectively, reconfirming induction of CD154 expression by these peptides (Fig. 2). Cells from Ad-GFP-immunized mice and naive mice, which were stimulated with a mixture of peptides C, D, and J, yielded an enrichment rate of 14.6 and 12.4, respectively.
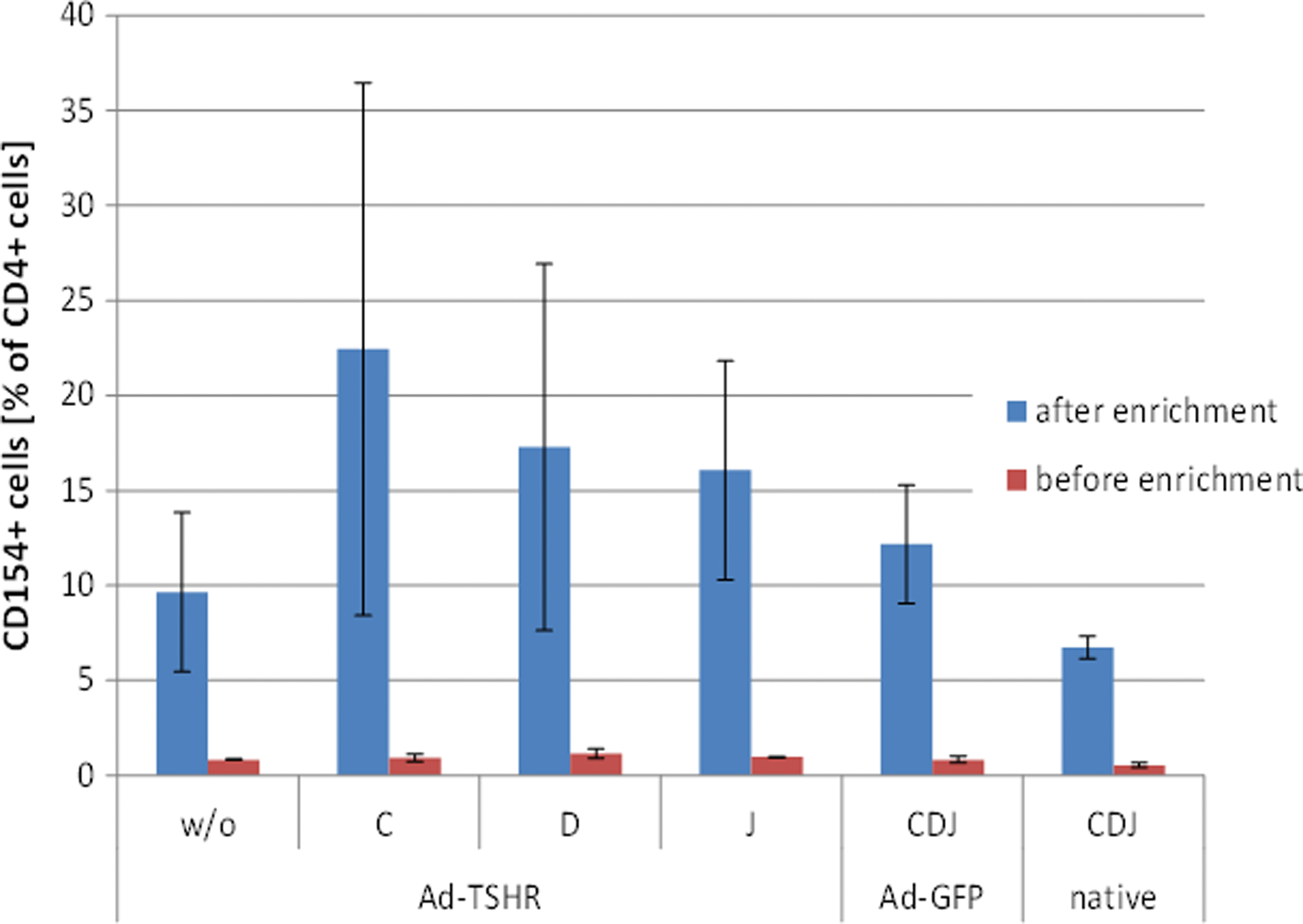
Identification of CD154-expressing T helper cells before and after enrichment. Activated spleen-derived cells were pooled and enriched using an anti-CD154-PE antibody and anti-PE MicroBeads after o/n stimulation with peptide or PMA/ionomycin-positive control compounds. Cells were analyzed for CD154 expression before and after enrichment. The bars show CD154-stained cells as percentage of all CD4-stained T helper lymphocytes (means of four independent samples ± SEM). PE, phycoerythrin; PMA, phorbol mystirate acetate. Color images are available online.
Enriched cells were subsequently cultivated for 6 days in murine T cell medium in the presence of 50 ng/mL IL-15. Harvested cells were analyzed for binding of TSHR-specific MHC II tetramers labeled with the fluorescence dye APC and co-staining for CD4. Binding of peptide C-MHCII tetramers numerically increased in Ad-TSHR-immunized mouse samples, although this result narrowly failed to reach significance. In contrast, numbers of CD4-positive lymphocytes stained with peptide D- and J-MHCII tetramers were similar to those stained with class II associated invariant peptide (CLIP) MHC II tetramer. Hardly, any background staining was observed in Ad-GFP-immunized mice and naive control mice (Fig. 3).

Detection of antigen-specific CD4-positive T cells by immunotype H2-IAd MHC II tetramers. Isolated spleen cells of Ad-TSHR-immunized BALB/c mice were stimulated with TSHR-derived peptides C, D, or J or left unstimulated (“control”). Ad-GFP or native control mice were stimulated with mixtures of all three peptides. Cells were enriched for CD154-expressing cells, expanded for six days in culture, and were subsequently analyzed using MHC II tetramers with bound C, D, or J peptides. Clip peptide was used as negative control tetramer. (
HLA-DR3-transgenic Bl/6 mouse splenic lymphocytes
The presence of HLA-DR antigens was confirmed in ear clips of 14 randomly selected mice by genotyping. All investigated pups and animals of the HLA-DR3-transgenic line showed clearly detectable HLA-DR3 gene expression by (semi-)quantitative PCR, with no detectable variation between individual animals or different litters—no findings below threshold of detection were observed in these animals. Expression of HLA-DR on blood cells was further confirmed by FACS in all analyzed mice. In TSHR protein-immunized HLA-DR3-transgenic mice, peptide 10 had been identified as a high-affinity epitope, and peptide 37 had been previously described to elicit antigen-specific immune responses (19,24). Peptides I and E had been identified as epitopes in Ad-TSHR-immunized DR3-transgenic mice by Pichurin et al. (18). Table 4B summarizes the sequences and hTSHR locations of the respective peptides.
Analysis of CD8-depleted cells by MHC II tetramer staining
Unspecific staining of CD8-positive cells using DR3 tetramers was observed in previous experiments. A CD154-based enrichment was attempted but did not succeed in HLA-DR3-transgenic mice. Therefore, to avoid capture of tetramers by CD8 cells, a depletion step using MACS MS columns and labeling of cells with CD8 MicroBeads were included in the protocol for detection of TSHR-specific CD4 cells. Cells reduced in the CD8 population were first treated with dasatinib to inhibit phosphorylation-dependent downregulation of T cell receptors (TCRs) (25) and then incubated with tetramers at 37°C for 1 hour before staining of T cell markers CD3 and CD4. HLA-DR3-tetramers loaded with p10 identified antigen-specific T cells from Ad-TSHR-immunized HLA-DR3-transgenic mice compared with CLIP control HLA-DR3-tetramers (Fig. 4B) and also compared with the results with cells from Ad-GFP-immunized control mice, although CLIP control tetramer produced relatively strong unspecific staining compared with the control without tetramer. In contrast, HLA-DR3-tetramers coupled to pE nearly missed significance compared with CLIP control DR3-tetramers (Fig. 4B).

HLA-DR3-MHCII tetramer signals on splenic T lymphocytes. Splenic T lymphocytes were isolated from Ad-TSHR-immunized HLA-DR3 transgenic mice. The figure shows results of six experiments with tetramers, which were prepared to contain the TSHR-derived specific peptides p10, p37, pE, and pI versus the nonspecific CLIP tetramer control. (
Human PBMCs
Gating strategies
Representative density blots of the different B and T cell populations are shown. They illustrate the gating strategies for the detection of viable B cells, naive, and memory B cells (Supplementary Fig. S1), as well as for T cells, cytotoxic T cells, and T helper cells (Supplementary Fig. S2).
Gating strategy for Treg cells
Human Treg cells are often characterized on the basis of expression of the cell surface markers CD3, CD4, CD25, CD127, and of the transcription factor FoxP3. Because CD3 was omitted as a general T cell marker, using an anti-CD14 antibody separately, it was shown that CD4+CD14+ cells are excluded from the lymphocyte gate, which is separated from the monocyte gate by lower values for forward and side scatter (Supplementary Fig. S3).
A representative example for gating of Treg cells using CD4, CD25, CD127, and FoxP3 as markers is shown in Supplementary Figure S4.
Effect of freezing and thawing on the viabilities of PBMCs
All thawing procedures of cryopreserved PBMCs yielded very similar results. The viabilities of all samples were >83%, which can be considered as generally high (Table 5).
Mean Viabilities of Peripheral Blood Mononuclear Cells and Lymphocytes After Thawing
Viabilities were analyzed in flow cytometry with FVS780 and FVS520 in eight independent samples. Mean values are shown with standard deviations.
PBMCs, peripheral blood mononuclear cells.
Quantification of basic B and T cell populations in human PBMCs by flow cytometry
To quantify B cells, memory B cells, naïve B cells, and T cells, in addition to the viability stain, several B cell markers were added to the cells. The mean quantities of B cell populations are shown in Table 6A, and of T cells (markers CD3, CD4, and CD8) in Table 6B.
B Cell (A) and T Cell (B) Populations
Mean values of eight samples are shown with standard deviations.
Miyahira 2012 (35).
Product flyer of Leucosept (Greiner Bio-One International GmbH, Kremsmünster, Austria).
Stone et al. (31).
ThermoFisher Scientific (36).
Direct comparison of B and T cell populations before and after freezing and thawing
T cell populations were analyzed from four, B cell populations from six different donors. Freezing and thawing had no major impact on the quantities of the main T lymphocyte populations, that is, after thawing, the mean T helper and cytotoxic T cell population sizes were 99% to 103% of that before freezing, the maximal difference seen with one donor was 13%.
Similarly, the mean proportions of B cells and naive B cells did not decline upon freezing, whereas the proportion of memory B cells slightly decreased compared with existing references (Table 6A). CD19-positive B cells were recovered by 72% to 181%, depending on donor. The detected population of naive B cells tended to increase in size, representing between 102% and 207% of those detected before freezing. In contrast, the relative size of memory B cells decreased in frozen cells compared with fresh cells, making up 53% to 94% of fresh cells.
Comparison of the population sizes of fresh and frozen cells expressing Treg markers
Population sizes of CD4-positive cells expressing only one of the relevant markers for Treg cells were analyzed in PBMCs of five healthy donors and compared between fresh cells and frozen cells after thawing. CD4-positive cells were identified in a histogram. The second marker, for example, CD25, was then identified in another histogram applied to CD4-positive cells only.
The portion of CD25-expressing cells varied between 28.5% and 46.0% of CD4+ cells in fresh PBMCs (Fig. 5A), with a recovery rate after freezing/thawing of 82.2% to 102.7%. CD127-expressing cells varied between 50% and 65% in fresh PBMCs and were significantly higher in frozen compared with fresh cells (relative % frozen vs. fresh: 102.6–120.4%). The population of FoxP3-expressing cells varied between 4.2% and 8.5% in fresh cells (Fig. 5A) and did not vary: the number of FoxP3+ in frozen was 95.1% compared with nominally 114.0% in fresh PBMCs—the figure of 114% corresponds to 100% recovery and some variation in detection.

Comparison of cell populations expressing relevant markers for Treg cells (
Comparison of the portion of Treg cells in human PBMCs before and after freezing
Treg cells were identified by gating CD4-positive T cells with high expression of the CD25 and low or no expression of CD127, with concomitant expression of the nuclear transcription factor FoxP3. In freshly isolated PBMC, a Treg cell portion of 5.06% ± 1.02% (% of CD4-positive cells) was found (Fig. 5B) and of 4.71% ± 1.0% after freezing/thawing.
MHC II tetramer staining of Graves' patients' T lymphocytes
HLA-DR3-tetramers loaded with p10 identified antigen-specific T cells from HLA-DR3-positive patients with Graves' disease, as shown in Table 7: A specific signal of p10-MHCII tetramer was observed in CD4+ pMHCII+ cells, whereas the negative controls (Clip-MHCII tetramer, non-CD4 cells, or healthy volunteers) yielded signals at background level. In contrast, all other tested MHC II tetramers did not result in specific signals in patients' PBMCs.
Results of Specific Tetramer Stainings with Peripheral Blood Mononuclear Cell from Patients with Graves’ Disease
HLA-DR3-tetramers loaded with p10 identified antigen-specific T cells from HLA-DR3-positive patients with Graves' disease. Results are mean values from three individual patients, which were determined in duplicates, with SEM.
All comparisons of p10 tetramer versus controls yielded p-values <0.05.
SEM, standard error of the means.
These results are comparable to those obtained with splenic lymphocytes from HLA-DR3-transgenic mice (Fig. 4)—but the nonspecific background was lower in patient cells.
Comparison of cytokine levels in supernatants of fresh or recently frozen PBMCs
PBMCs before and after a freeze/thaw cycle were nonspecifically stimulated with LPS, a component of the bacterial cell wall, or Zymosan, exhibited on the surface of yeast cells, or antibodies stimulating T cells via the TCR complex and co-receptor. After overnight incubation, supernatants were stored frozen and tested for secretion of proinflammatory cytokines IL-1beta, TNF-alpha, and IL-6 by sandwich ELISA. Very strong signals for IL-1beta or IL-6 were obtained in Zymosan- or LPS-treated PBMCs irrespective of using fresh or recently frozen cells. The TNF-alpha response in Zymosan-treated PBMCs was somewhat lower in frozen compared with fresh cells but nevertheless high enough to clearly recognize an immune response compared with unstimulated control cells (Fig. 6). Stimulation of T cells with anti CD3 and CD28 antibodies revealed elevated but lower levels of IL-6 and TNF-alpha compared with Zymosan-treated cells indicating that T cells are not the main players in an acute inflammatory event.
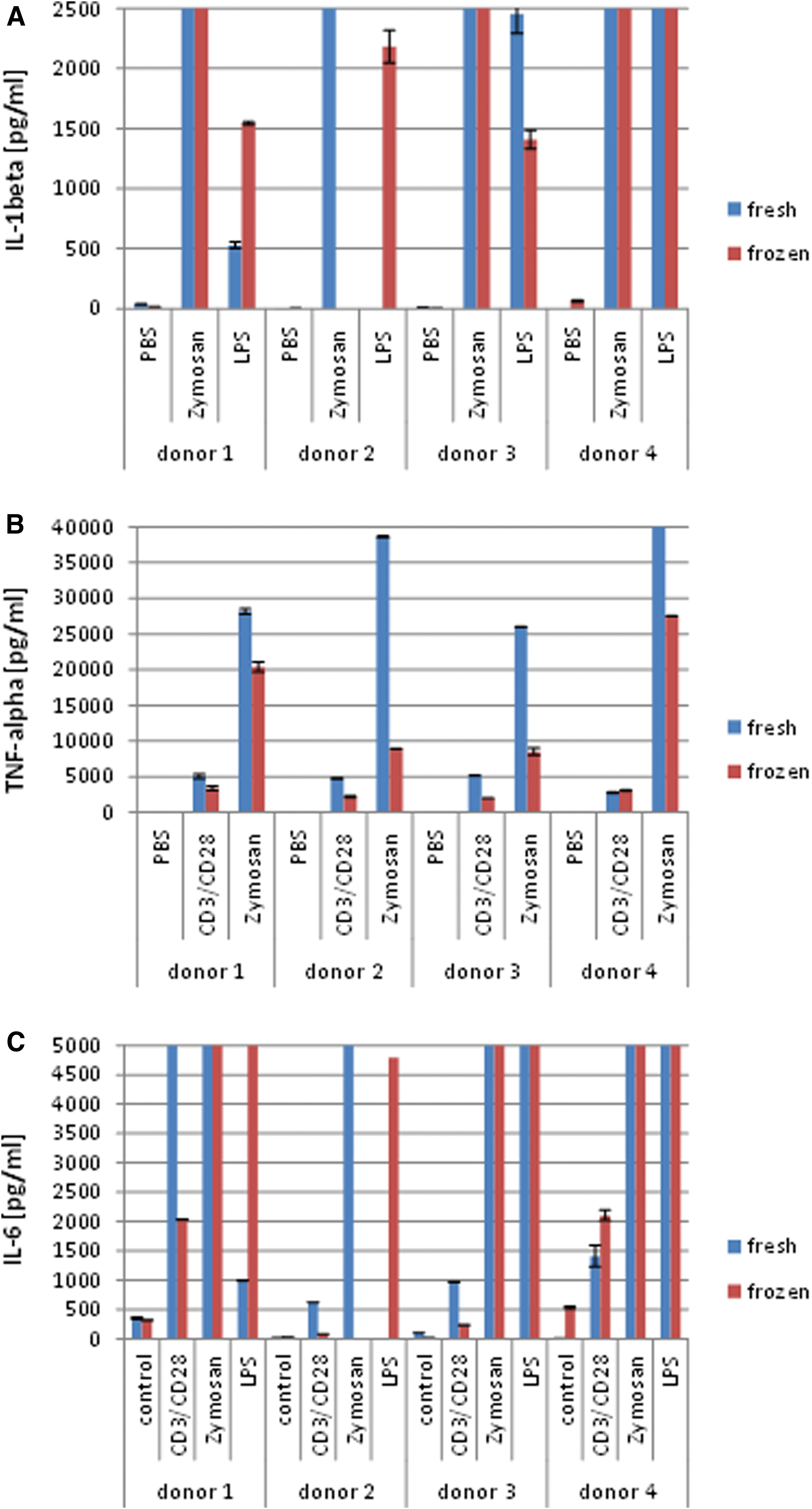
Cytokine levels in supernatants of stimulated human PBMCs measured by enzyme-linked immunosorbent assay. Freshly isolated or thawed PBMCs were stimulated with 1 μg/mL anti-CD3 antibody in combination with 5 μg/mL anti-CD28 antibody, 10 μg/mL Zymosan, or 100 ng/mL LPS. The bars show mean values with SD (
Discussion
The experiments described here were performed to detect antigen-specific T lymphocytes in mouse models of Graves' disease and in sera from patients with Graves' disease using MHC II tetramers.
Peptides C, D, and J were selected from the literature as being able to stimulate IFN-γ secretion from splenic lymphatic cells of Ad-TSHR-immunized BALB/c mice (11). Peptides C and J, but not peptide D, resulted in increased co-expression of intracellular IFN-γ and CD154 in stimulated splenic lymphatic cells of long-term Ad-TSHR-immunized mice. For peptide C, but not peptide J, this result was corroborated by MHC II tetramers coupled to this specific peptide, which resulted in relevant tetramer staining of CD154-enriched spleen lymphocytes from Ad-TSHR-immunized mice. These measurements can be used to support future therapeutic studies in mouse models, such as on the effect of fingolimod to mitigate Graves' orbitopathy by altering T lymphocyte subsets (26).
The signal obtained with peptide J-tetramer was only slightly above background. Prediction algorithms on binding of peptide J to I-Ad of BALB/c mice by MHC binding yielded a sixfold higher affinity of peptide J compared with peptide D (27). Thus, weaker binding occurred despite of higher binding prediction. It may possibly be due to lower binding avidity of J-tetramer for TCRs, which may be too weak to yield a stable labeling of T cells, whereas during stimulation of T cells by antigen-presenting cells, multiple MHC/TCR complexes stabilize this binding and thus enable stimulation. This would be in line with the observations by others (28), who found that detection of T cells with tetramers is limited to high-affinity pMHCII/TCR interactions, which comprise only 5–30% of the total naive antigenic repertoire. However, the prediction algorithms especially on MHC II binding are not very accurate, and this might be another example illustrating this issue.
In addition, we were also able to establish a HLA-DR3-tetramer assay, which detected antigen-specific T lymphocytes in splenic lymphocytes of HLA-DR3-transgenic mice. Also in these mice, just one of the specific epitopes described in previous references (18,19) worked for the respective tetramer assay. As expected, antigen-specific peptides differ between the mouse H2-IAd and the human HLA-DR3 immunotype background. The same p10-loaded MHC II tetramer was also used to identify antigen-specific cells in thawed human PBMCs.
Moreover, our findings are in line but do not completely reconfirm the results of Kirchhoff et al. (29) that in immunized BALB/c mice all cytokine-producing T helper cells were positive for CD154, and of McDyer et al. (30) who reported that induction of IFN-γ is dependent on CD40L/CD40 stimulation in PHA-stimulated human PBMCs.
This study also describes and evaluates a standardized protocol for freezing and thawing of human PBMC samples, which had almost no effect on the population sizes of CD3-positive T cells, T helper cells, and cytotoxic T cells. Similarly, the mean proportions of B cells and naive B cells did not decline upon freezing, whereas the proportion of memory B cells slightly decreased (about 20%).
Under the experimental conditions described here, for most samples analyzed, cell viabilities determined with FVS780 were ∼95%
Freezing of PBMCs, storage at −80°C, and thawing were found to have no significant impact on the percentage of Treg cells as reported by Van Hemelen et al. (33), although the marker CD127 was detected on a higher percentage of cells after freezing and thawing. Because Treg cells are characterized by the absence or only low expression of CD127, this alteration had obviously no influence on Treg cell identification. In this study, the mean portion of Treg cells among CD4-positive cells was 5.06%
To measure specific Treg cells, the gating method posted on the web page of Excyte Expert Cytometry in 2016 by Snyder-Cappione (34) was used with the modification of omitting the T cell marker CD3. Instead, we analyzed CD4/CD14 staining in advance and verified that these double positive cells are not present in the lymphocyte gate, thus confirming that CD4-positive cells in the lymphocyte gate represent T cells. Within the T4 cell subtype sample, TSHR-specific cells were identified with the assay that had been built up using HLA-DR-transgenic mice.
Thawed human PBMCs were also suitable to test peptides or biologics for a proinflammatory effect on immune cells by measuring cytokines such as IL-1beta, TNF-alpha, or IL-6 in the supernatant of the cells after overnight incubation. The assay is easy to perform and shows sensitive detection of these cytokines in unstimulated cells and a strong response after endotoxin treatment. Additional investigation of human stimulated T cell subsets or intrathyroidal T cells will add relevant information on the results of the present study and will focus on the identification of TSHR-specific T cells in immunized mice and human Graves' patients, measuring such T cells in a time-dependent manner.
In conclusion, we have established protocols which enable to identify TSHR-specific T lymphocytes in hyperthyroid murine models with murine and human T cell epitopes, and in sera from patients with Graves' disease. The described optimized and standardized protocol for cryopreserving PBMCs offers the perspective of blood sampling from patients and long-term PBMC storage in future studies. Moreover, we have standardized detection of lymphocyte cell subpopulations and cytokine release from heparin- or EDTA-treated thawed human blood samples.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4