
Abstract
Background:
Loss-of-function mutations of thyrotropin receptor (TSHR) are one of the main causes of congenital hypothyroidism. As for many disease-associated G-protein coupled receptors (GPCRs), these mutations often affect the correct trafficking and maturation of the receptor, thus impairing the expression on the cell surface. Several retained GPCR mutants are able to effectively bind their ligands and to transduce signals when they are forced to the cell surface by degradation inhibition or by treatment with chaperones. Despite the large number of well-characterized retained TSHR mutants, no attempts have been made for rescue. Further, little is known about TSHR degradation pathways. We hypothesize that, similar to other GPCRs, TSHR retained mutants may be at least partially functional if their maturation and membrane expression is facilitated by chaperones or degradation inhibitors.
Methods:
We performed in silico predictions of the functionality of known TSHR variants and compared the results with available in vitro data. Western blot, confocal microscopy, enzyme-linked immunosorbent assays, and dual luciferase assays were used to investigate the effects of degradation pathways inhibition and of chemical chaperone treatments on TSHR variants' maturation and functionality.
Results:
We found a high discordance rate between in silico predictions and in vitro data for retained TSHR variants, a fact indicative of a conserved potential to initiate signal transduction if these mutants were expressed on the cell surface. We show experimentally that some maturation defective TSHR mutants are able to effectively transduce Gs/cAMP signaling if their maturation and expression are enhanced by using chemical chaperones. Further, through the characterization of the intracellular retained p.N432D variant, we provide new insights on the TSHR degradation mechanism, as our results suggest that aggregation-prone mutant can be directed toward the autophagosomal pathway instead of the canonical proteasome system.
Conclusions:
Our study reveals alternative pathways for TSHR degradation. Retained TSHR variants can be functional when expressed on the cell surface membrane, thus opening the possibility of further studies on the pharmacological modulation of TSHR expression and functionality in patients in whom TSHR signaling is disrupted.
Introduction
Loss-of-function (LOF) mutations of thyrotropin receptor (TSHR) are one of the principal causes of congenital hypothyroidism (1,2). TSHR is a G-protein coupled receptor (GPCR) that is characterized by a seven-transmembrane alpha-helix B-subunit and an extracellular A-subunit, linked by disulfide bonds (3,4).
TSHR post-translational modifications are required for correct trafficking, maturation, and activity (3,5,6). The N-linked glycosylation, happening in the endoplasmic reticulum (ER), is fundamental (Fig. 1A). Acquisition of mannose-type carbohydrates permits the interactions with molecular chaperones (Fig. 1B), required for correct receptor folding, homodimerization, passage through the ER quality control system, and translocation to cis-Golgi (Fig. 1C) (3,5,7). In trans-Golgi, TSHR finally acquires the complex-type carbohydrates (Fig. 1D) that characterize the mature form expressed on the cell surface and undergoes tyrosine sulfation, which is fundamental for activation (6,8). On the plasma membrane, TSHR is cleaved by an unidentified enzyme with loss of a short sequence of variable size, peptide C (Fig. 1E). The receptor is finally composed by an extracellular A-subunit and a transmembrane B-subunit linked by disulfide bonds (3,5,9,10).

TSHR maturation and the hypothesized degradation pathways. THSR comprises a seven-transmembrane alpha helix B-subunit (green) and an extracellular A-subunit (purple). It is synthesized in the ER as a full-length unglycosylated protein [(
For many disease-associated GPCRs, including TSHR, LOF is most often due to poor cell surface expression, rather than from intrinsic deficiencies in signal transduction. The abnormal mutant conformation leads to interactions with alternative molecular chaperones (3,11,12), ER blockage, and degradation by proteasome or autophagosome (Fig. 1G, H) (13,14). Different retained GPCR mutants are able to effectively bind their ligands and transduce intracellular signals when forced to the cell surface (15 –17). The use of chemical chaperones is a well-explored area to overcome ER retention of various membrane receptors (18 –20). Currently, little is known about TSHR degradation pathways and no attempts have been made to rescue TSHR mutants.
The aim of our work was to better elucidate TSHR degradation pathways and the possibility of TSHR mutants being rescued with chemical chaperones. We concentrated our attention on two different mutants that we previously described: the TSHR p.N432D, which is retained in the ER as a high-mannose form, and the p.P449L, which is usually expressed on the plasma membrane but has impaired signaling (21). We then validate our findings in two other retained TSHR mutants (22,23). Our results show for the first time that maturation-defective TSHR mutants are able to transduce Gs/cAMP signaling when rescued by the chemical chaperone Trimethylamine-N-oxide (TMAO). Moreover, we provide new insights on the TSHR degradation mechanism, as our results suggest that aggregation-prone mutants are directed toward the autophagosomal pathway instead of the canonical proteasome system.
Materials and Methods
Patients
Patients bearing the p.N432D and p.P449L variants were previously described (21). The study had been approved by the local ethics committee (code: 05C002_2010). Informed written consent for genetic analyses had been obtained.
Chemicals
Cell culture reagents, ProLong Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole, LysoTracker Red DND-99, ER-Tracker Green, Alexa-Fluor conjugated and HRP-conjugated antibodies, and Restore Western blot Stripping reagent were purchased from Thermo-Fisher (Waltham, MA). Mouse anti-Actin Ab-5 was purchased from BD (Franklin Lakes, NJ). Anti-TSHR antibodies BA8 (Cat#SC_BA8, RRID:AB_2716681), 3G4 (Cat#SC_3G4, RRID:AB_2716682), and 28.1 (Cat#SC_28.1, RRID:AB_2716683) were a gift of Dr. S. Costagliola (Free University of Brussels, Belgium) (24 –28). Anti-E-Cadherin antibody was purchased from Abcam (Cambridge, United Kingdom), and anti-VDAC antibody was purchased from Santa Cruz (Dallas, TX). bTSH, Anti-green fluorescent protein antibody, TMAO, dimethyl sulfoxide, and MTT were purchased from Sigma-Aldrich (St. Louis, MO).
In silico prediction
The TSHR variants membrane expression and functionality was assessed through the TSHR mutation database (29). Fifty-five variants were subjected to in silico predictions and assigned as damaging or not damaging as specified in Supplementary Methods.
Cell culture, transfection, treatments, and viability assay
COS-7 cells were grown in Dulbecco's modified Eagle medium (Thermo-Fisher) supplemented with 10% fetal bovine serum and penicillin–streptomycin (Sigma-Aldrich). TSHR expression vectors were previously described (21). pSVL plasmids containing wild type (WT), p.E34K, and p.R46P TSHR variants were a gift of Dr. Tonacchera (University of Pisa, Italy) (22,23).
Transfection, degradation modulation, and rescue were performed as described in Supplementary Methods. Cell viability was tested by using the MTT assay (30).
Western blotting
Cells were lysed in sodium dodecyl sulfate (SDS) buffer (62.5 mM Tris-HCl pH 6.8, 2% SDS) supplemented with protease, phosphatase, and proteasome inhibitors. Membrane preparations were obtained with Plasma Membrane Protein Extraction Kit (Abcam) following the manufacturer's instructions. Total Cellular Membranes and Plasma Membranes fractions were then processed as described in Supplementary Methods. Band intensity was quantified with ImageJ software (31).
Immunofluorescence and confocal microscopy
Samples were processed as previously described (32), and a detailed description is in Supplementary Methods. Images were acquired with EclipseTi-E inverted microscope with IMA10 × Argon-ion laser System by Melles Griot; images were acquired with CFI Plan Apo VC 60 × Oil (Nikon).
Flow cytometry
Samples were processed as previously described (32), and a detailed description is in Supplementary Methods. Measurements were performed with an FACSCalibur flow cytometer (BD Biosciences). Data were analyzed with Flowing Software 2.
Functional assays
cAMP pathway activity was assessed with Cignal CRE Reporter (luc) Kit (Qiagen, Hilden, Germany), while Gq11/IP3 pathway activity was measured with IP-One ELISA assay kit (Cisbio, Waltham, MA), following the manufacturer's instructions, as described in Supplementary Methods.
Statistical analyses
All experiments were independently repeated at least three times, as indicated in the figure legends. After normal distribution and variance similarity evaluation, two-sided unpaired t-test (eventual Welch's correction for groups with different variances), one-way analysis of variance with Bonferroni post hoc test, Kruskal-Wallis H test with Dunn's post hoc test, and Chi-square test were used as indicated in the figure legends.
For concentration–effect curves of Gs/cAMP signaling, a log(agonist) versus normalized response–variable slope equation was used for curve interpolation and parameters definition.
For confocal experiments, the degree of colocalization was quantified through Pearson's correlation coefficient, as measured with Nikon NIS Elements software. Correlation was defined as strong for a Pearson's correlation coefficient higher than 0.8, moderate when higher than 0.5 but <0.8, and weak when higher than 0.2 but <0.5. In all figures, data are shown as mean ± standard error of the mean. Data were analyzed by using GraphPad Prism 5 software, and significance was expressed as p values (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
In silico prediction and in vitro data of receptor functionality are significantly discordant in retained mutants
We obtained complete information about in vitro functionality and subcellular localization of 55 LOF TSHR variants (29) and categorized them as intracellular-retained or membrane-expressed (Supplementary Table S1). These mutations were subjected to in silico predictions and assigned as functional or nonfunctional.
The comparison of in vitro and in silico data revealed a significantly higher discordance rate among the retained group (12/24 and 7/31 mutants with positive prediction but in vitro LOF for intracellular-retained and membrane-expressed, respectively, p = 0.0471) (Table 1). Such a discrepancy may indicate that some ER-retained TSHR mutants can potentially transduce signals if expressed on the cell surface. We, thus, explored the degradation mechanisms and chaperone rescue on two previously reported (21) TSHR LOF variants: the intracellular-retained p.N432D and the membrane-expressed p.P449L.
In Vitro and In Silico Functionality Concordance of Thyrotropin Receptor Variant with Different Subcellular Localization
Distribution of TSHR variants from in vitro data and in silico predictions concordance of function in relation to subcellular localization. For 55 different variants, data about in vitro subcellular localization and functionality were obtained through literature review. In silico prediction were obtained with six different online tools (polyphen-2, PROVEAN, SIFT, PhD-SNP, PANTHER, and SNPs&GO) and TSHR variants were then assigned as damaging (more than 50% of predictions were concordant as functionally damaging or impaired functionality) or not damaging (50% or more of predictions were concordant as neutral or benign), as described in Supplementary Methods and in Supplementary Table S1.
Each variant was then assigned to one of the four groups: membrane localization with in vitro and in silico concordance on functionality, membrane localization with in vitro and in silico discordance on functionality, intracellular retainment with in vitro and in silico concordance on functionality, and intracellular retainment with in vitro and in silico discordance on functionality.
TSHR, thyrotropin receptor.
N432D variant is arrested in the ER and forms different aggregates
We performed confocal microscopy with two different antibodies: the BA8 directed against a conformational epitope on the mature A-subunit and the 3G4 raised against a linear epitope in the C-peptide that recognizes principally immature forms (24,25).
WT TSHR and p.P449L variant had a normal membrane expression in all transfected cells (Fig. 2A), whereas p.N432D had a variable pattern detected by BA8 antibody, with three main morphologies: small intracellular aggregates (SA), perinuclear signal (PS), and cytoplasmic macroaggregate (MA) (Fig. 2B). SA and PS were the most frequent ones, while in around 10% p.N432D pattern had the characteristics of more than one morphology (mixed morphology) (Fig. 2B). In contrast, p.N432D variant staining with 3G4 antibody revealed a constant pattern of diffuse PS that was not detectable with BA8 antibody (Fig. 2B). This difference may indicate the presence of a significant amount of immature or incorrectly folded receptors recognized only by the 3G4 antibody but not by BA8 (24). Interestingly, SA were similar to the puncta that characterize misfolded GPCR mutants degraded by autophagocytosis (33,34), while MA were suggestive of perinuclear aggregates related to the proteasome degradation pathway (34,35).
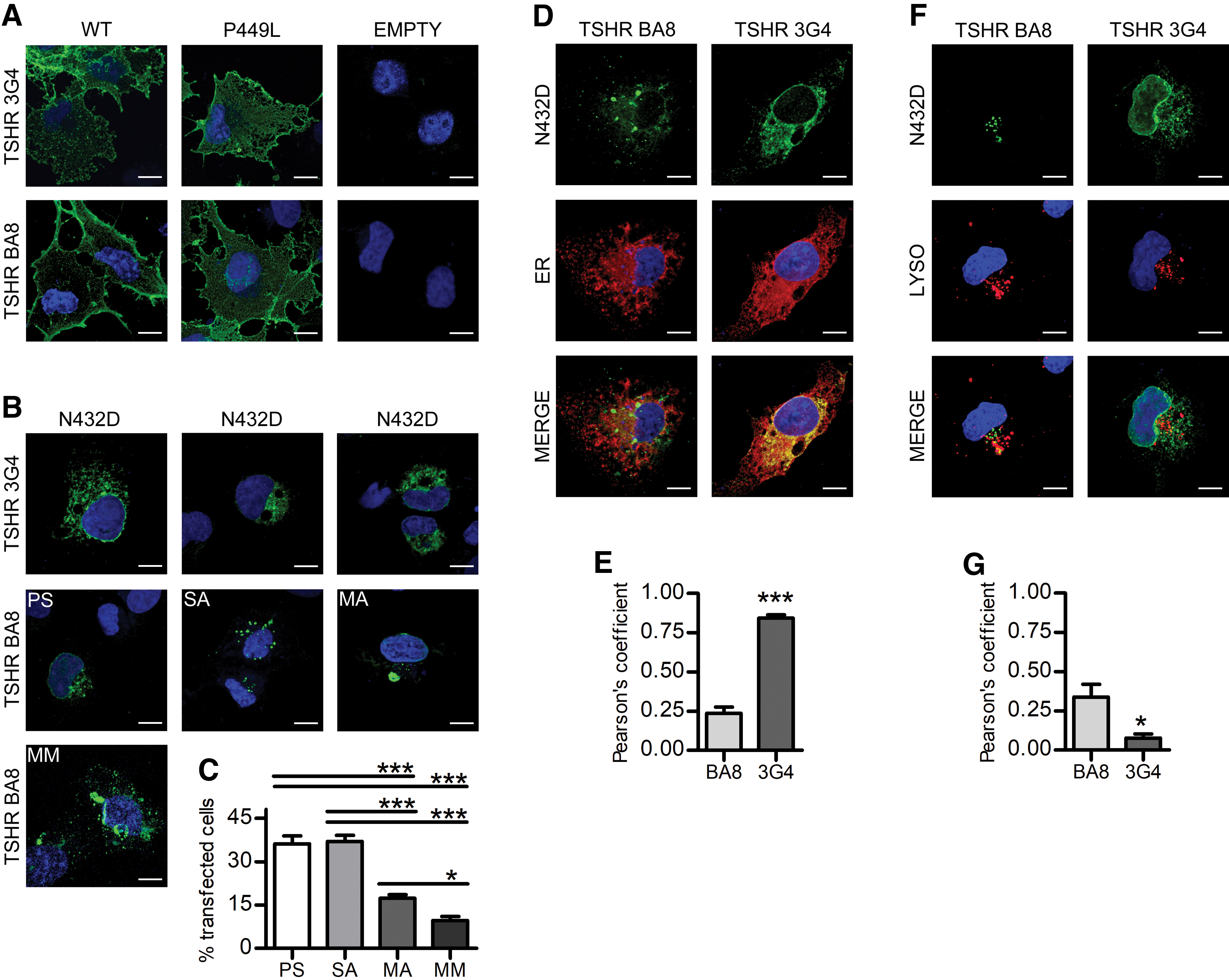
p.N432D mutant is retained in the ER and lysosomes in different aggregates. (
Co-staining with p.N432D variant and ER or late endosome/lysosome markers showed that the majority of the protein recognized by 3G4 antibody was, indeed, localized in the ER (Fig. 2D, E). Different features of ER stress, such as vacuoles and enlarged morphology (36), were also detected in transfected cells. On the other hand, the aggregates recognized by the BA8 antibody showed a mild co-localization with endosomes/lysosomes (Fig. 2F, G).
TSHR mutants are degraded through different pathways
For many GPCRs, the ubiquitin-proteasome system is the main degradation system (37,38), while mutants prone to form aggregates are directed toward autophagic degradation (33,39). We evaluated whether that was the case by performing Western blot analysis in different conditions, with the 28.1 antibody that recognizes the full-length receptor at different stages of maturation and the cleaved A-subunit (28,40).
MG132 proteasome inhibitor induced a significant accumulation of mature WT TSHR and p.P449L variant, confirming the fundamental role of this pathway. However, only a strong accumulation of the immature form was detected for p.N432D (Fig. 3A, B). NH4Cl, an autophagocytosis inhibitor, did not cause significant alterations in the total WT TSHR, although a change in the amount of mature form could be appreciated, as previously reported (5). On the other hand, endolysosomal inhibition induced a more effective accumulation of p.P449L and of immature p.N432D than the proteasomal inhibitor (Fig. 3A, B).

WT and mutant TSHRs are degraded through different pathways. (
Confocal microscopy experiments revealed a significant increase in SA after autophagocytosis inhibition, while a significant increase in MA was seen after proteasome inhibition; concomitant inhibition had an intermediate effect (Fig. 3C), thus confirming the Western blot data.
Autophagocytosis activation with LiCl induced an almost complete degradation of the p.N432D variant, with milder effects on p.P449L and no effects on WT (Fig. 3D, E). Moreover, only p.N432D expression induces JNK 1/2 phosphorylation, an event linked to autophagocytosis activation (41) and significantly reduced cell viability (Supplementary Fig. S1A, B), thus confirming the role of autophagocytosis in misfolded TSHR degradation.
The chemical chaperone TMAO restores p.N432D mutant membrane expression
The p.N432D mutant does not maturate even if protein degradation is inhibited, but as immature TSHR can signal when expressed on the plasma membrane (42,43), we investigated whether this was the case with the use of different chemical chaperones.
Western blot experiments showed that, unlike in other GPCRs (44), treatment with glycerol was not effective in the p.N432D variant rescuing (Supplementary Fig. S3). Nevertheless, TMAO treatment (45) increased the maturation of all TSHR variants, but with major effects on p.N432D whose A-subunit intensity reaches levels similar to the WT control, indicating possible membrane expression; the high-mannose form had a larger increase in the WT and p.P449L TSHR (Fig. 4A, B). Hybridization with 3G4 antibody revealed that TMAO treatment caused a significant increase in a high-molecular-weight band (around 200 kDa) (Supplementary Fig. S2A, B) that has been identified as dimers of high-mannose forms (43), whose formation was fundamental for the passage through ER quality control.
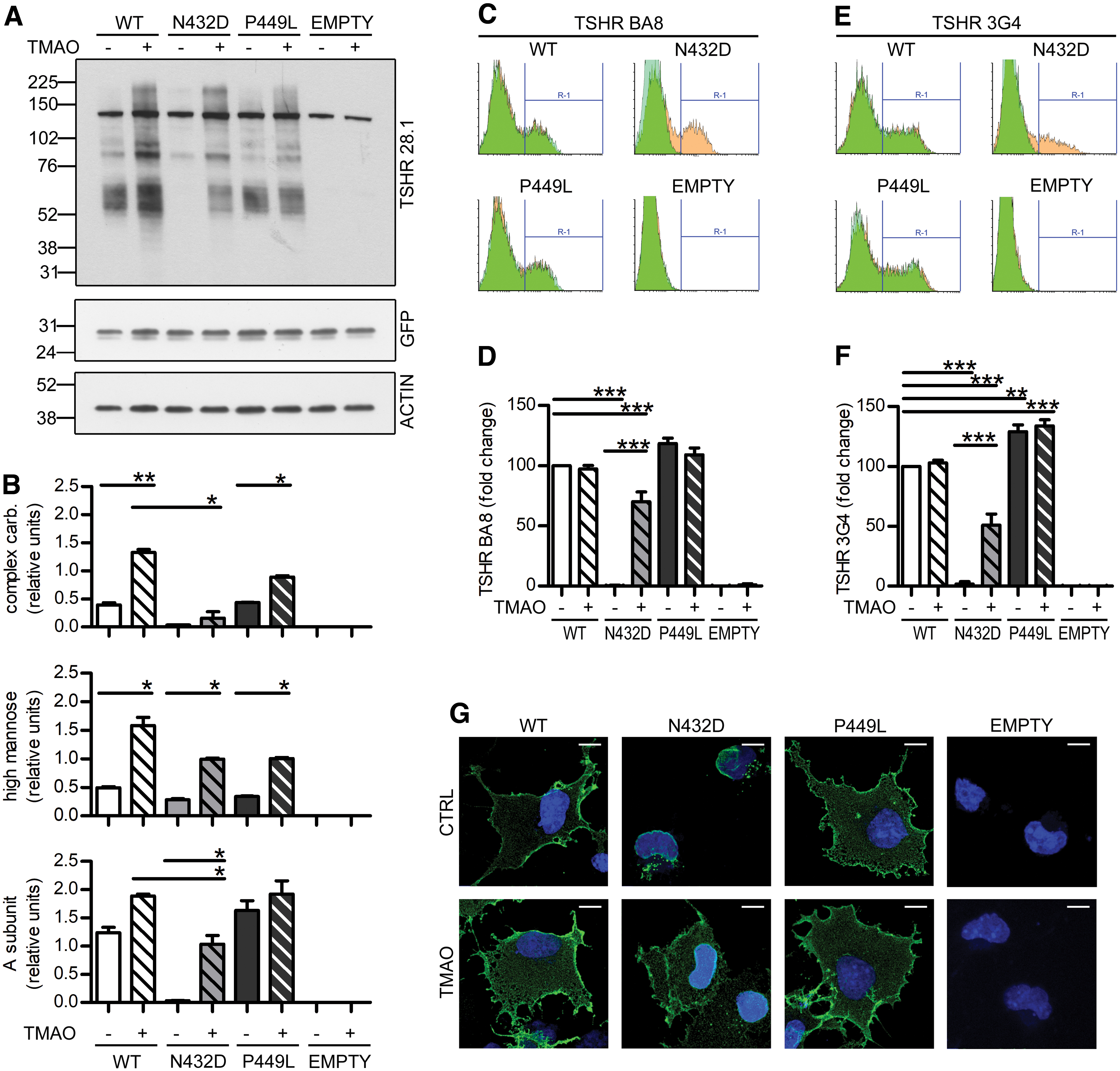
TMAO restores p.N432D mutant trafficking and membrane expression. (
Fluorescence-activated cell sorting experiments in non-permeabilized cells confirmed cell-surface expression of TMAO-treated p.N432D. Interestingly, membrane expression resulted in 75% of the WT control with the BA8 antibody (Fig. 4C, D), and only around 50% of the WT control with the 3G4 antibody (Fig. 4E, F) with a BA8:3G4 ratio of 1.62 ± 0.25 (p < 0.05 vs. treated WT), a finding that indicated increased cleavage of the p.N432D mutant (25).
Immunofluorescence experiments with BA8 staining confirm p.N432D membrane expression after TMAO treatment. The increased intracellular staining for TMAO-treated TSHRs is in agreement with the increased immature forms detected in Western blot experiments (Fig. 4G).
Cellular membrane fractionations revealed that the A-subunit was the predominant form on the cell surface. Moreover, TMAO promoted WT TSHR translocation on the cell surface, as we detected decreased levels of all TSHR maturation forms in the total membrane extracts and increased levels in the plasma membrane extracts. This effect was not seen in the p.P449L variant, which was also insensitive to TMAO effects (Supplementary Fig. S2C, D).
Membrane expression uncovers the functional potential of p.N432D and other retained variants
The evaluation of rescued p.N432D variant signaling transduction abilities through Gs/cAMP and Gq11/IP3 pathways revealed that, indeed, the mutant was partially functional when expressed on the plasma membrane. In fact, although the Gq11/IP3 pathway remained greatly compromised (Fig. 5A), the maximal Gs/cAMP response was almost completely rescued (Fig. 5B). Concentration–effect curves (Fig. 5C and Supplementary Fig. S4A; Table 2) showed that TMAO treatment had virtually no effect on the Gs/cAMP signaling of either the WT or p.P449L mutant receptor, while the TMAO-treated p.N432D curve was right-shifted, indicating higher EC50 values.

TMAO treatment unveiled partial functionality of N432D and other known retained mutants. (
Membrane Expression and Functional Parameters of the Loss-of-Function Thyrotropin Variants
The table summarizes the main characteristics of the LOF THSR variants. Membrane expression was examined with flow cytometry as in Figure 4C and E. Maximal stimulation and EC50 were obtained from the experiment as in Figure 5. Values are expressed as mean ± SD.
Statistical analysis: Statistical significance was determined with one-way ANOVA. * p < 0.05, ** p < 0.01, *** p < 0.001 versus WT TSHR, §§ p < 0.01, §§§ p < 0.001 versus the respective untreated TSHR variant.
LOF, loss of function; SD, standard deviation; WT, wild type; TMAO, trimethylamine-N-oxide.
As a last step, we investigated two additional retained variants that had discordant in silico and in vitro functionality: the p.E34K, which has a reported membrane expression of 30% of WT, and the p.R46P, which is reported to be almost totally retained and with a very low ability to signal through the cAMP pathway (22,23).
TMAO treatment induced an increase in the cleaved A-subunit levels in the p.E34K variant and greatly enhanced the maturation of the retained p.R46P one (Fig. 5E, D), with effects similar to the ones observed in p.N432D. Accordingly, functional assays revealed a significant increase in both Gs/cAMP and Gq11/IP3 pathways for p.E34K and a significant rescue of the signaling abilities of p.R46P (Fig. 5F, G), with concentration–effect curves and EC50 similar to those of the WT (Fig. 5H).
Discussion
In the present work, we reveal two important issues regarding the possible intracellular destiny of the folding-defective TSHR mutants. First, they may be degraded not only through the proteasomal pathway but also through an alternative autophagosomal-like pathway that kicks in as an emergency exit after retention in the ER. Second, they can at least partially function if forced to the cell surface by using chemical chaperones. Our data provide a possible explanation for the observed lack of concordance between the in silico prediction of receptor functionality and in vitro findings, as misfolded mutants that retain signaling abilities may have a premature maturation arrest, intracellular retention, and subsequent degradation.
The involvement of the lysosomal system in the degradation of misfolded TSHR mutants is a new and interesting finding. In particular, p.N432D has such structural changes that prevent it from passing the ER quality control. In the ER, the mutant is likely to form aggregates (SA) that cannot be retro-translocated to the cytoplasm where the proteasome system operates, but are instead degraded by alternative autophagocytosis (Figs. 1–3 and Supplementary Fig. S1), a mechanism similar to the one previously described for gonadotropin-releasing hormone receptor mutant p.E90K (33).
The TMAO treatment likely inhibits the formation of ER aggregates while promoting receptor homodimerization, sheltering p.N432D from ER quality control and allowing advancement to the Golgi compartment and finally to the plasma membrane (3,46), as also indicated by the appearance of the A-subunit bands in plasma membrane preparations. Nevertheless, its maturation does not seem to follow regular steps even after TMAO treatment as we detected very low levels of complex carbohydrates form (Fig. 4A, B).
There are two possible explanations for this. The first and most likely hypothesis is that only a small percentage of plasma membrane p.N432D mutant reach full maturation, while most of it is still blocked at the high-mannose stage. Membrane expression of immature TSHR has already been described (26,42,43,47), and TSHR with reduced glycosylation sites has TSH binding affinity and EC50 for cAMP that are indistinguishable from the mature one (3). In this case, the p.N432D maturation limiting factor may be the ability to form dimers in the ER compartment, as the staining with 3G4 antibody showed a significant increase in the levels of immature TSHR dimers after TMAO treatment (Supplementary Fig. S2A, B). The increased cleavage indicated by the variation in BA8:3G4 ratio (Fig. 4C, E; Table 2) can then be explained by the already known higher sensitivity to protease action of immature TSHR (25,42).
The second possible explanation is that the TMAO-treated p.N432D mutant reaches full maturation, but all the mature receptors undergo proteolytic cleavage and thus only the A-subunit is detected. This may be explained by an increased sensitivity of the TSHR mutant to proteases or because a lower amount of mutant TSHR on the membrane is more effectively processed by proteases.
Irrespective of these considerations, functional assays show that p.N432D mutant is able to bind TSH and signal when expressed on the plasma membrane (Fig. 5A–C; Table 2). The lack of Gq11/IP3 pathway activity may be explained by the intrinsic differences between Gs and Gq interaction with TSHR.
First of all, the Gq11/IP3 pathway is more dependent on the total amount of cleaved receptor (10) and on TSHR homodimerization abilities (48), and TMAO-treated p.N432D has an absolute amount of cleaved receptor present on the plasma membrane that is definitely lower than WT (Fig. 4 and Supplementary Fig. S2).
In addition, interactions between TSHR and Gq are more demanding than the ones with Gs (4,49), and an in silico model predicted that p.N432D mutation results in more severe modifications that can affect the interaction with G-proteins (21). The TMAO treatment can either mask these conformational alterations or force the mutant through a more correct conformation, enough to achieve a partial rescue of Gs interactions and cAMP signaling but not enough to restore the more demanding interactions with Gq.
These speculations are also supported by the findings in two other discordant mutants, where TMAO treatment more efficiently rescued Gs/cAMP than the Gq11/IP3 signaling (Fig. 5D–H and Supplementary Fig. S4B).
In conclusion, TSHR can be degraded through proteasome or autophagosome pathways depending on specific structural defects. The chaperone TMAO allows TSHR mutants to pass ER quality control, increasing cell surface expression. As in other GPCR-related diseases, TSHR LOF mutations mainly cause ER retention, as detected by the discrepancy between in silico predictions and in vitro data. As demonstrated here, retained mutants that are brought to the cell surface are able to effectively transduce intracellular signals. These findings open the possibility of further studies on pharmacological modulation of TSHR expression and functionality in patients with disrupted TSHR signaling.
Footnotes
Authors' Contributions
E.S.G., A.L., and V.G. designed and performed the experiments. V.V. and M.B. contributed to the experiment planning. L.P. supervised the experimental work and provided research funds. All authors contributed to the writing and revision of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The work was partially supported by Ricerca Corrente funds of Istituto Auxologico Italiano (project: EPIPOT; funding code: 05C002_2010). E.S.G. was partially supported by Fondazione Anna villa e Felice Rusconi (CF: 80008670129) PhD scholarship.
Supplementary Material
Supplementary Method
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1