
Abstract
Resistance to thyroid hormone alpha (RTHα) is caused by mutations in thyroid hormone receptor α (THRA). Little is known about the natural history and treatment of RTHα, and diagnosis before the age of 1 year has not been previously reported. A de novo heterozygous THRA mutation (pC380SfsX9) was identified in a 10-month-old female investigated for developmental delay, hypotonia, macrocephaly, and severe constipation. Treatment with levothyroxine was accompanied by an appropriate rise in thyroxine (T4), triiodothyronine (T3), as well as decrease in thyrotropin levels and in the T3/T4 ratio with a trend toward normalization of peripheral markers of thyroid hormone action. THRA pC380SfsX9 results in extreme RTHα.
Introduction
Resistance to thyroid hormone alpha (RTHα) is a rare inherited condition characterized by impaired sensitivity to thyroid hormone (TH) due to mutations in the thyroid hormone receptor α (THRA) gene. The clinical presentation of RTHα is variable and can be associated with abnormal neurologic development, congenital heart abnormalities, and abnormal growth. This corresponds to the tissues that are predominately TH receptor alpha (TRα) dependent. Serum thyroid function tests (TFTs) show a low to low normal thyroxine (T4) concentration and a normal to high triiodothyronine (T3) and thyrotropin (TSH). Thirty-eight subjects have been reported in 24 families with 24 different mutations in the THRA gene (references in Supplementary Data). The THRA gene generates several products by alternative splicing and use of internal promoters (1,2). The known mutations in the THRA gene consist of missense or frameshift mutations that alter the ligand-binding domain of the receptor, altering TRα1 ability to bind to T3 as well as coactivators (SRC-1) and corepressors (NCoR).
Methods
Details are provided in the Supplementary Data.
Case Report
The proposita was the second child of healthy parents both of Ashkenazi Jewish decent born at 41 weeks gestation by normal vaginal delivery. Her newborn thyroid screen was normal. She presented with an umbilical hernia, Stahl's ears, and large fetal size (birth length 53.08 cm, 98th centile, and weight 3.49 kg, 75th centile). Due to decreased O2 saturation and labored breathing, she was placed in the neonatal intensive care unit for 10 days, requiring continuous positive airway pressure support. A heart murmur prompted an echocardiogram that revealed mild supravalvular pulmonary stenosis with an atrial septal defect. A borderline prolonged QT interval was noted as well. At four months, concern of developmental delay was supported by hypotonia, poor weight gain, and severe constipation. A nevus of the right cheek presented at 6 months of age. Together these clinical findings led to genetic testing. A de novo heterozygous THRA gene mutation was identified (c1139delG; pC380SfsX9), affecting only the α1 subunit of the TRα protein. TFTs at 10 months of age presented an elevated T3/T4 ratio, and low total T4 levels (Supplementary Table S1). Levothyroxine (LT4) treatment for 10 months produced an appropriate rise in total T4, total T3, and free T3 levels as well as a suppressed TSH (<0.01 mU/L). However, the T3/T4 ratio decreased from 46 at baseline to 31 (Supplementary Table S1). Peripheral tissue markers of TH action demonstrated an expected decrease of serum cholesterol from 310 to 100 mmol/L and creatine kinase that decreased from 297 to 70 IU/L. Paradoxically, sex hormone binding globulin and ferritin levels also decreased 67% and 22%, respectively. As previously reported in other patients with RTHα, the child had significant normocytic anemia with a hemoglobin of 9.0 g/L, which did not change throughout the course of LT4 treatment (9.1 g/L at 20.8 months of age).
Despite the high levels of serum T4 achieved, awake and sleeping heart rate remained 100–110 beats per minute. Owlet® home monitoring recorded 7 hours of “deep sleep” over several nights and oxygen saturation from 93% to 99% with an average of 96%. LT4 treatment markedly improved constipation with 1 bowel movement daily from one every 3–4 days before treatment. At 20.8 months of age, weight was at the 28th percentile while length was at <1st percentile (Supplementary Figs. S1 and S2); head circumference was 19.2 in (48.7 cm) at the 93rd percentile; the anterior fontanelle was 1.9 in (4.9 cm). The propositus was not talking, walking, nor able to stack 2 blocks at 19.6 months.
Discussion
The novel THRA mutation (c11139delG; pC380SfsX9) selectively impairs the function of TRα1. Clinical and biochemical characteristics of reported patients with RTHα were examined (Fig. 1A–C). Considering the presence of TRα in heart, gut, bone, and parts of the brain, RTH manifests in these tissues and is noted to be more prevalent in the group with mutations affecting only the TRα1 protein isoform. A discussion of the different THRA mutations and clinical manifestations of the disease giving insight into pathogenesis and treatment is given in the Supplementary Data.
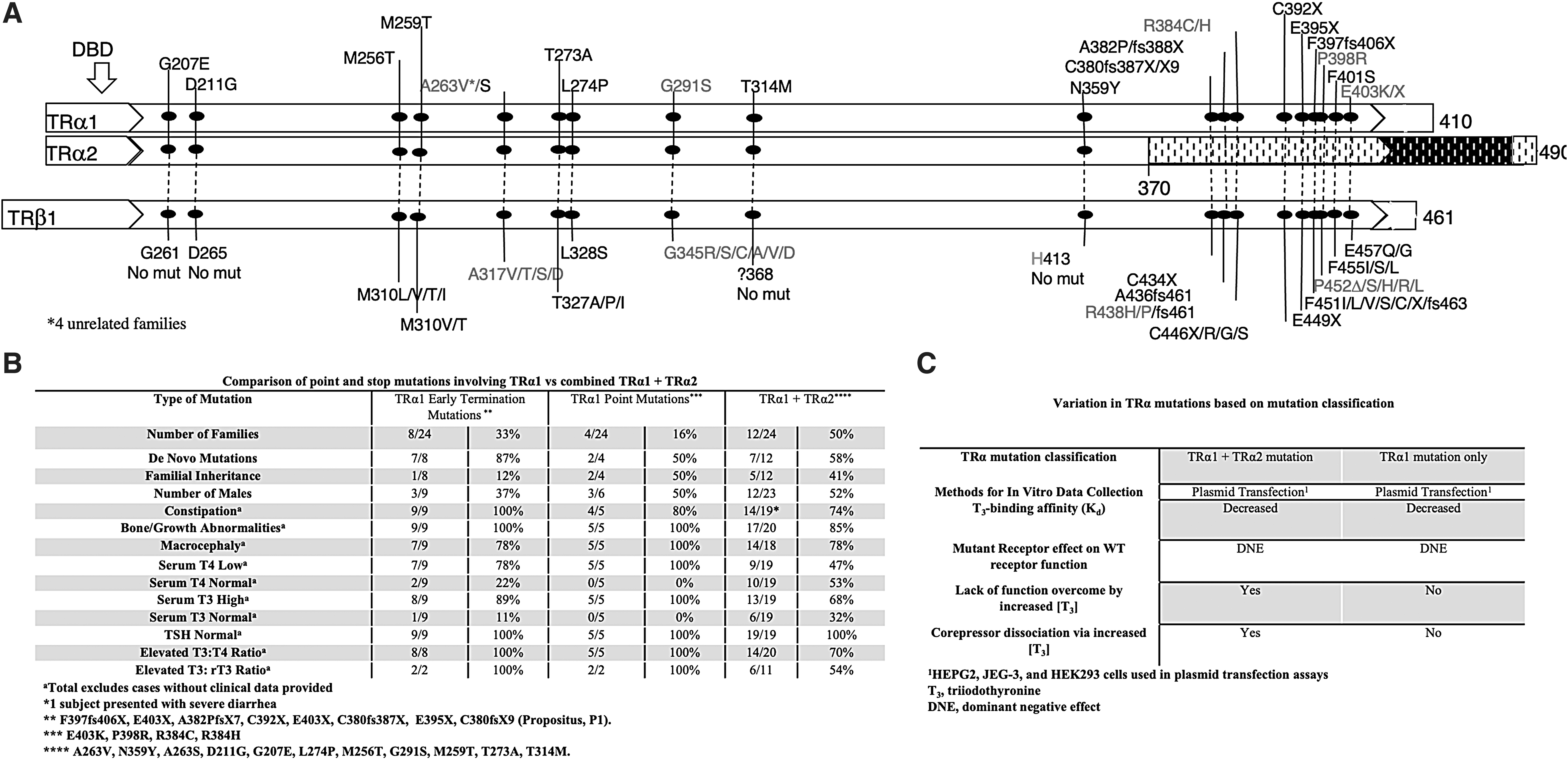
TH metabolism is regulated by the three deiodinase selenoenzymes (D) and their distinct roles and regulation are to be considered when exploring potential mechanisms for the serum TFTs observed in this defect. D1 is regulated primarily by TRβ and is an activating enzyme, resulting in increased conversion of T4 to T3, while it also metabolizes reverse T3 (rT3). D3 is regulated by TRα (3) and is an inactivating enzyme as it degrades the active hormone T3, and converts T4 to the inactive metabolite rT3. Therefore, in the presence of impaired TRα and subsequent impaired D3 activity, T3 is not adequately degraded and its concentration increases. The resulting elevated T3 potentially increases the activity of the liver D1, resulting in the lower serum T4 and the relatively high T3. The low D3 and high D1 activities also potentially explain the low rT3 observed in this defect. Of note, rT3 levels are in the normal range even when serum T4 reaches very high levels during LT4 treatment (Fig. 1A). It is striking that a similar constellation of serum TFTs is seen in patients with MCT8 deficiency where there is an increase in T3 at the expense of T4 with normal TSH (4).
The improvements with early LT4 treatment have been modest, and despite early aggressive treatment, bone and neurologic development have not normalized. As the long-term effects of high LT4 doses in the context of such a deleterious THRA mutation are unknown, we will continue to closely monitor the patient and adjust the regimen, taking into consideration all developmental parameters. It is notable the profound postnatal growth delay compared with the apparently normal intrauterine growth based on the length at birth (Supplementary Fig. S2), potentially indicating that other mechanisms might be able to compensate in utero. These data suggest that future therapies would require an analogue that will bind the mutant receptor in the cytoplasm to free the wild-type TRα or the delivery of an small interfering RNA that would specifically degrade the mutant TRα.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the National Institutes of Health, USA, DK15070 to S.R. and DK110322 to A.M.D., and by funds from the Esformes Thyroid Research Fund to R.E.W.
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1