
Abstract
Background:
Pituitary resistance to thyroid hormone (PRTH) is often seen in congenital hypothyroidism (CH), presenting as elevated thyrotropin (TSH) values despite (high-)normal thyroid hormone (TH) values achieved by levothyroxine treatment. In this study, we describe a girl with CH who was referred because of difficulties interpreting thyroid function tests. She was thought to have PRTH associated with CH, but genetic studies discovered a pathogenic variant in THRB, causing resistance to TH (RTH-β).
Methods:
Clinical, genetic, and biochemical data of the proband's family were collected.
Results:
The 3-year-old girl was diagnosed with CH due to a homozygous pathogenic c.470del p.(Asn157Thrfs*3) SLC5A5 variant in the neonatal period. She needed a notably high levothyroxine dose to normalize TSH, leading to high free thyroxine levels. There were no signs of hyperthyroidism. Sequencing identified a heterozygous pathogenic c.947G>A p.(Arg316His) THRB variant.
Conclusions:
To our knowledge, this is the first report of concomitant SLC5A5 and THRB variants causing CH and RTH-β.
Introduction
Defects in the sodium-iodide symporter (NIS) are a rare cause of primary congenital hypothyroidism (CH). NIS is mainly found at the basolateral membrane of thyroid follicular cells, where it enables the first step of thyroid hormone (TH) synthesis by actively transporting iodide (I−) into thyroid follicular cells. Homozygous and compound heterozygous pathogenic variants in the NIS gene SLC5A5 (OMIM No. 601843) result in thyroid dyshormonogenesis (1,2). Depending on the nature of the SLC5A5 variant, patients' phenotypes may vary from severe hypothyroidism to a compensated euthyroid state. Treatment consists of levothyroxine and/or iodide supplementation to prevent goiter and hypothyroidism.
In general, the main goal in the treatment of primary CH is to normalize thyrotropin (TSH) levels. In the presence of a functioning hypothalamic–pituitary–thyroid (HPT) axis, a normal TSH level is considered to reflect a normal TH status. However, in severe forms of CH, normalization of TSH levels can only be achieved by establishing TH levels above the age-specific reference interval (RI) (3 –5). This phenomenon is postulated to be the result of an altered HPT-axis set-point with some degree of pituitary resistance to TH (PRTH), probably caused by fetal hypothyroidism affecting HPT-axis maturation (3,4). The altered set-point presents a conundrum for the physician as TSH values cannot reliably be used to assess the adequacy of levothyroxine supplementation. It is recommended to maintain free thyroxine (fT4) levels within the RI, preferably in the upper quartile, and accept TSH values just above the RI (4).
In this report, we describe a 3-year-old girl who was diagnosed in the neonatal period with thyroidal hypothyroidism due to a homozygous pathogenic SLC5A5 variant. TSH values could only be normalized with a high levothyroxine dosage effectuating remarkably high fT4 values, which was initially attributed to an altered TSH-fT4 set-point. However, further genetic testing revealed a heterozygous pathogenic THRB (OMIM No. 190160) variant causing RTH-β, explaining high TSH values in combination with normal fT4 values. To our knowledge, this is the first report of a concurrent NIS defect and RTH-β.
Materials and Methods
Patients
We studied a family of Turkish origin, including a mother, father, two sons, and one daughter (the proband). The proband and her family agreed to participate and signed informed consent forms. Blood samples were obtained for thyroid function tests and genotyping. This study was approved by the medical ethical committee of Amsterdam UMC, location AMC (METC AMC), project number W20_138.
Mutational analysis
All coding exons and intron/exon borders of the DUOX2, DUOXA2, FOXE1, HOXA3, NKX2-1, SLC26A4, TPST2, TG, IYD, TPO, TSHR, and SLC5A5 genes were sequenced in 2014 using the Sanger technique. In 2018, all coding exons and intron/exon borders of the THRB gene were sequenced by Sanger sequencing as well. The pathogenicity of identified variants was predicted using Alamut Visual® software (Interactive Biosoftware, Rouen, France).
Case presentation
The proband, a 3-year-old girl, was the third born child of consanguineous Turkish parents (fourth degree consanguinity; parents are first cousins). She was born at term after an uneventful pregnancy. In the first week of life, she was admitted to the pediatric ward because of mild neonatal hyperbilirubinemia and poor feeding. Blood samples taken on the seventh day of life demonstrated severe primary CH, with a low fT4 [4.0 pmol/L; RI 20.5–37.1 pmol/L (6)], an elevated TSH [>100 mU/L; RI 1.0–8.4 mU/L (6)] (both ECLIA Roche Diagnostics) and a strongly elevated thyroglobulin (1560 pmol/L; RI 0–60 pmol/L) (Brahms Thermo Scientific). A thyroid ultrasound showed a normally positioned but enlarged thyroid gland with decreased echogenicity. Thyroid scintigraphy showed no uptake of 123Iodine in the head/neck region.
Mutational analysis of SLC5A5
A eutopic thyroid gland without any uptake of 123Iodine is highly suggestive for a NIS defect—in the absence of maternal anti-TSH receptor antibodies. Sanger sequencing of DUOX2, DUOXA2, FOXE1, HOXA3, NKX2-1, SLC26A4, TPST2, TG, IYD, TPO, TSHR, and SLC5A5 revealed a previously reported homozygous pathogenic SLC5A5 (Chr19:NM_000453.2) c.470del p.(Asn157Thrfs*3) variant (7), predicted to elicit nonsense-mediated messenger RNA decay. This confirmed the diagnosis of CH due to an NIS defect.
Follow-up
Levothyroxine treatment (31.25 μg) was instituted and fT4 levels were within the normal range after 12 days (Fig. 1). Feeding normalized, and hyperbilirubinemia was successfully treated with phototherapy. However, since the start of treatment the patient needed a notably high dose of levothyroxine to normalize TSH. fT4 values were high at every routine investigation, which led to think the patient received multiple doses of levothyroxine in anticipation of the doctor's visit and laboratory test (Fig. 1). Despite this her parents persistently claimed to give medication as prescribed. Because of these puzzling thyroid function tests and a relatively high levothyroxine dose (7 μg/kg/day), she was referred to the department of paediatric endocrinology of the Amsterdam University Medical Center for further investigation.
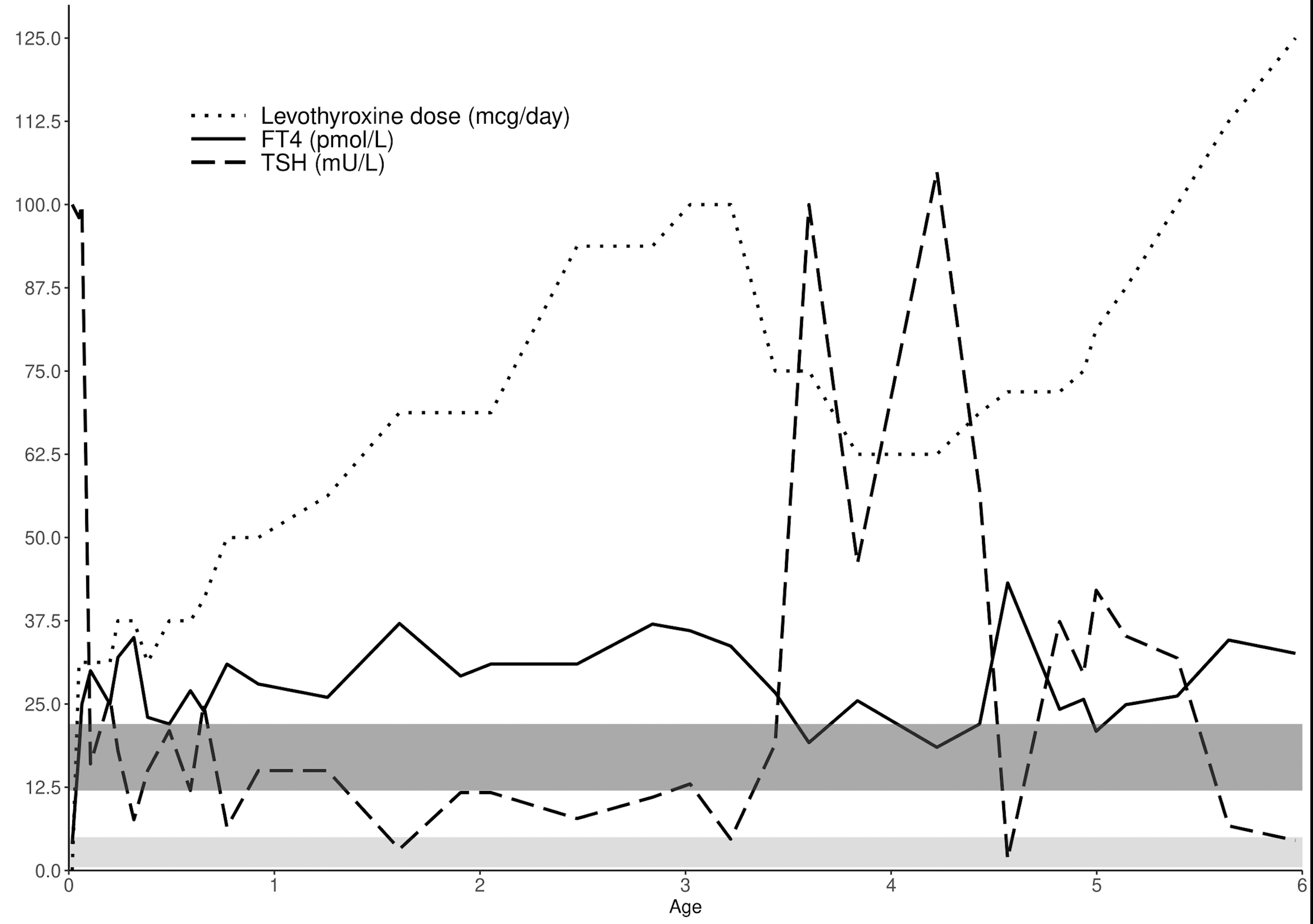
Thyroid function test results of the proband over time. fT4 and TSH were measured by immunoassay (ECLIA, Roche Diagnostics). The dark shaded area represents the adult fT4 RI and the light shaded area represents the adult TSH RI. fT4, free thyroxine; RI, reference interval; TSH, thyrotropin.
At that time parents reported a normal motor and cognitive development, although receptive and expressive language development were delayed, for which the girl was receiving speech therapy. There were no clinical signs of hyper- or hypothyroidism. Height was 95 cm (−1.0 standard deviation score [SDS]; target height −1.6 SDS), weight 15.9 kg (weight for height +1.4 SDS). Heart rate and blood pressure were normal. The thyroid gland was mildly enlarged at ultrasonography, with a total volume of 3.7 mL (normal age-specific volume 2.5 ± 0.75 mL). At laboratory evaluation, which was carried out under levothyroxine therapy, fT4 was 33.7 pmol/L (RI 12–22 pmol/L), total thyroxin 270 nmol/L (RI 70–150 nmol/L) (in-house radioimmunoassay), free triiodothyronine (fT3) 7.3 pmol/L (RI 2.9–6.6 pmol/L) (Architect, Abbott Diagnostics), and TSH was normal, 4.7 mU/L (RI 0.5–5 mU/L). The following investigations were performed to asses peripheral tissue TH sufficiency: total serum cholesterol was 4.51 mmol/L (RI <5 mmol/L), sex hormone-binding globulin (SHBG) was 100 nmol/L (RI 14–114 nmol/L) (Architect, Abbott Diagnostics) and the resting energy expenditure (REE) was 55 kCal/kg/day, with an expected REE according to Schofield's formula of 53.9 kCal/kg/day. In case of peripheral tissue thyrotoxicosis one would expect low cholesterol, elevated SHBG and an elevated REE. Thus, the proband's normal test results indicated that peripheral tissues were probably in a euthyroid state. Still, the levothyroxine dose was tapered to guard brain development for possible negative effects of high fT4 values. fT4 was brought within normal limits (18.5 pmol/L) resulting in a strongly elevated TSH (105 mU/L). At this point, there were still no obvious complaints or clinical signs of hypothyroidism. Assessment of skeletal age later showed a significant delay (4.2 years at a calendar age of 6.5 years).
Mutational analysis of THRB
Congenital TH resistance was considered as an explanation for the strongly elevated TSH and Sanger sequencing of the thyroid hormone receptor-beta (THR-β) gene THRB (Chr3:NM_000461.4) was performed. A heterozygous c.947G>A p.(Arg316His) (R316H) variant was identified, which was interpreted as pathogenic based on prior reports (8 –10). Together with the findings of previous tests (nonsuppressed TSH, elevated fT4, and no clinical or biochemical signs of thyrotoxicosis), this confirmed the diagnosis of generalized RTH-β. Taking RTH-β into account, the levothyroxine dose was again increased until TSH levels returned to within its normal limits at an fT4 concentration of ∼35 pmol/L at age 5 years (Fig. 1).
Segregation analysis
The proband's mother carried the same heterozygous R316H variant although she did not have an RTH-β phenotype. She received levothyroxine treatment for subclinical hypothyroidism (fT4 14.3 pmol/L [RI 10.3–24.5 pmol/L], TSH 7.5 mU/L [RI 0.4–4 mU/L]) since pregnancy of her first child at age 25 years. Her thyroid function test results were normal (Fig. 2). Incomplete penetrance has been associated with the R316H variant before (8). The proband's two elder brothers were healthy and had normal neonatal CH screening results. Genetic testing showed both boys carried the R316H variant (Fig. 2). Thyroid function tests were suggestive of compensated RTH-β (Fig. 2). They had no signs of hyperthyroidism at physical examination and their thyroid glands were not enlarged at palpation and ultrasonography. The c.470del p.(Asn157Thrfs*3) SLC5A5 variant was present in a heterozygous state in the proband's parents and brothers (Fig. 2).

Pedigree and chromatograms. First diagnostic fT4 and TSH values at referral are presented in the table. fT4 (RI 12–22 pmol/L) and TSH (RI 0.5–5 mU/L) were measured by immunoassay (ECLIA, Roche Diagnostics). The chromatograms depict the SLC5A5 and THRB variants as found in the proband. This figure clearly demonstrates the lack of a genotype–phenotype correlation: while both brothers of the proband have mildly elevated fT4 levels, their mother is unaffected by RTH-β. The proband (indicated with a black arrow) has highly elevated fT4 levels upon normalization of TSH with levothyroxine treatment, possibly due to a combination of RTH-β and RTH as a result of primary CH. †Blood draw during treatment with levothyroxine of the mother and proband was, respectively, 50 and 100 μg once daily. CH, congenital hypothyroidism; RTH-β, resistance to thyroid hormone.
Discussion
We report on a patient with CH due to an NIS defect and concurrent generalized RTH-β. A supraphysiological levothyroxine dose was necessary to maintain TSH within normal ranges. At first, laboratory tests seemed suggestive for noncompliance to levothyroxine treatment or CH-associated altered HPT-axis set-point. However, careful interpretation of additional laboratory tests hinted at RTH-β.
In children with severe CH there is often an altered TSH-fT4 set-point with persistent mildly elevated TSH, when fT4 is kept within the RI (5). However, the observed relation between fT4 and TSH in the proband could not be confidently explained by this phenomenon, as it was more exaggerated than observed in other CH patients (4). At first, levothyroxine dose was tapered because of high fT4 values at follow-up (11). This led to a sharp rise in TSH to >100 mU/L. The levothyroxine dose was increased after RTH-β was genetically confirmed, aiming at normalization of TSH and accepting elevated fT4 levels. The proband did not develop clinical or biochemical signs of hyperthyroidism indicating generalized RTH-β.
This report is not the first report of hypothyroidism alongside RTH-β, as it has been established before in autoimmune hypothyroidism, CH due to ectopic lingual thyroid and CH due to thyroid dysgenesis (12 –15). The previously published cases of CH and RTH-β involve very similar initial biochemical analyses and follow-up. To our knowledge, this is the first case in which SLC5A5 and THRB genetic variants cause CH in combination with generalized RTH-β.
THR-β is a nuclear hormone receptor that mediates transcription of various genes. There are two isoforms of THR-β as a result of alternative splicing, THR-β1 and THR-β2. THR-β2 is expressed in hypothalamus and pituitary, thus mediating central effects of THs (16). For example, triiodothyronine (T3)-activated THR-β2 inhibits TSH-releasing hormone (TRH) promotor activity (17). R316H prevents this, stimulating TRH production and subsequent inappropriate secretion of TSH and TH. This may lead to signs associated with hyperthyroidism at the target tissues (expressing THR-β1 and/or THR-α). In the proband, R316H also affects THR-β1, which explains the generalized RTH-β phenotype.
Arginine at position 316 is located in the highly conserved C-terminal ligand binding domain, specifically nearby the T3-binding arginine residue at position 320 (18). The previously presented cases of CH and RTH-β also involve pathogenic variants in one of these arginine residues (R320H and R316C, respectively) (13,15). In silico studies indicated that R316H destabilizes critical structural elements and impairs hormone binding (18). This is congruent with T3 binding affinity studies of R316H THR-β, which show severely compromised hormone binding (Ka mutant protein/Ka wild-type protein = 0.02 ± 0.02) (19). Furthermore, THR-β forms heterodimers with retinoid X receptors (RXRs). RXR increases the transcriptional activity of THR-β by attracting cofactors (20). However, R316H elicits an impairment of thyroid receptor corepressor and coactivator binding (15,20). THR-β-RXR dimers normally greatly repress target gene expression in the absence of T3, leading to clinical signs associated with hypothyroidism. Thus, R316H possibly beneficially modulated fetal brain development in the proband since R316H impeded transcriptional repression.
Pathogenic variants in THRB constitute the most common form of congenital TH resistance (21,22). Numerous RTH-β case reports evidence considerable phenotypical heterogeneity with varying symptoms, ranging from asymptomatic generalized RTH-β with goiter, to thyrotoxic symptoms arising from elevated THs in tissues predominantly expressing TH receptor-α (such as tachycardia, palpitations, or skeletal dysplasia) (21,22). The consistent (pathognomonic) phenotype of patients with RTH-β is increased serum fT4 and fT3 concentrations, and a nonsuppressed TSH. Patients may also present with uncompensated RTH-β, which is associated with growth faltering (23). This clinical feature is in agreement with expression of THR-β in chondrocytes during endochondral ossification (24). The proband likewise exhibited a delayed skeletal age due to prior failure to recognize RTH-β and insufficient treatment.
The R316H THR-β variant has been described before in the context of two distinct RTH-β phenotypes. The current proband exhibits generalized RTH-β, and prior reports either described generalized RTH-β or pituitary RTH-β (associated with peripheral tissue hyperthyroidism) (8 –10,25). One case report of a woman with generalized RTH-β and goiter due to the R316H variant describes an onset of clinical hyperthyroidism at age 33, after which total thyroidectomy was performed (10). Also, the patient of the first report of RTH-β due to the R316H variant presented with clinical hyperthyroidism at the age of 12 (8). These data implicate conversion from a generalized to pituitary RTH-β phenotype may be possible and should be monitored because of possible life-threatening morbidity.
In this report and another previously identified R316H RTH-β kindred, RTH-β is absent in variant carrying family members (8). This incomplete penetrance is possibly related to differential expression of THRB alleles or THR-β isoforms due to epigenetic modulators (26,27), or differences in dimerization properties with, for example, RXRs (15). Differential expression of THRB alleles or isoforms over time could also explain the fact that patients present with signs of peripheral tissue TH excess later in life (instead of a congenital onset), as previously mentioned.
In summary, we describe a case of primary CH due to an NIS defect with persisting abnormal thyroid function test under levothyroxine treatment. This report underlines an important lesson in the management of CH: genetic analyses are very insightful in case of exaggerated PRTH ascribed to CH, and the finding of a pathogenic THRB variant optimizes treatment. This report furthermore highlights the lack of a genotype/phenotype correlation in RTH-β.
Footnotes
Authors' Contributions
The first draft of the article was written by Peter Lauffer, and all authors commented on subsequent versions of the article. Hennie Bikker performed genomic analyses. The project was supervised by Nitash Zwaveling-Soonawala and Paul van Trotsenburg. All authors read and approved the final article and confirm authorship.
Acknowledgments
The authors are thankful for the participation of the proband and her family.
Statement of Ethics
The authors have no ethical conflicts to disclose. The proband and her family agreed to participate and signed informed consent forms. This study was approved by the medical ethics committee of Amsterdam UMC, location AMC (METC AMC), project number W20_138.
Author Disclosure Statement
No competing interests exist.
Funding Information
The authors received no funding for writing this article.