
Abstract
Background:
Epigenetic disorders play an important role in the pathogenesis of autoimmune thyroiditis (AIT). Therefore, the study of the possible role of DNA methylation in AIT is of great significance to explore the pathogenesis of AIT.
Methods:
From May 2019 to June 2019, whole blood samples were collected from 176 AIT patients and 176 controls from different water iodine levels in Shandong Province, China. We used the Illumina Methylation 850K BeadChip to determine significant differences in methylation status of genes and used the MethylTarget™ assay to verify the methylation level in 176 cases and 176 controls. The relative mRNA levels of genes were detected by quantitative real-time-polymerase chain reaction.
Results:
There were multiple differential methylation sites in the HLA-DPB1 and PDCD1LG2 genes between the case and control population with different water iodine levels. Some target regions of HLA-DPB1 and PDCD1LG2 genes were negatively correlated with relative mRNA expression in the case and control populations and with different water iodine levels.
Conclusions:
There is differential methylation status in genomic DNA in patients with AIT. The methylation patterns of HLA-DPB1 and PDCD1LG2 genes related to cell adhesion molecule pathway may be different based on different water iodine levels. HLA-DPB1 and PDCD1LG2 genes related to the cell adhesion molecules pathway may play a role in the development of AIT. This study is registered with Chinese Clinical Trial Registry,
Introduction
With the in-depth study of the molecular biological mechanism of thyroid diseases, increasing attention has been paid to the role of epigenetics in thyroid diseases. DNA methylation is an important epigenetic regulatory mechanism. However, the understanding of DNA methylation in autoimmune thyroiditis (AIT) is still very limited. Iodine plays a key role in the synthesis of thyroid hormones. Proper iodine status is essential for maintaining normal thyroid morphology and function (1). However, continuous iodine prevention and environmental iodine exposure may increase the risk of iodine excess. The relationship between iodine intake and AIT is complex, and the pathogenesis of AIT is not completely clear, but it is certain that genetic and environmental factors are involved in the development of AIT (2). Therefore, the purposes of this study was to explore whether different iodine levels and DNA methylation levels in susceptible genes are significantly associated with AIT.
Subjects and Methods
Subjects
The subjects in this study were from different water iodine areas in Shandong Province of China from May 2019 to June 2019 (3). The different water iodine areas were iodine-fortification areas (IF areas: median water iodine [MWI] <10 μg/L, supply of iodized salt since 1994, qualified iodized salt consumption rate >90%), iodine-adequate areas (IA areas: MWI 40–100 μg/L, supply of uniodized salt), and iodine-excess areas (IE areas: MWI >300 μg/L, supply of uniodized salt). According to the AIT case inclusion criteria, a total of 176 AIT cases and 176 healthy controls were collected. They were matched at 1:1 in their living area, age, sex, and body mass index (BMI). There were 89, 47, and 44 pairs subjects in IF, IA, and IE areas, respectively. Whole blood samples of all subjects were collected and stored at −80°C.
The following criteria were used to select and exclude subjects. First, subjects in the case group included only patients with TPOAb, TgAb, or double antibody positivity, excluding patients with hyperthyroidism and subclinical hyperthyroidism, were identified as AIT patients according to the diagnostic criteria of AIT disease (4). Second, the control group's inclusion criteria were as follows: (a) normal healthy people matched in the case group by age, sex, BMI, and place of residence; (b) no history of acute or chronic disease, no history of long-term use of hormones or drugs, no personal or family history of thyroid disease or other autoimmune diseases, and no pregnancy; (c) no goiter on physical examinations, negative for TPOAb and TgAb levels on laboratory evaluation, free triiodothyronine (fT3), free thyroxine (fT4), and thyrotropin (TSH) were normal, and thyroid ultrasound revealed no abnormalities (normal gland size, shape, internal echo, and no nodules).
This study was approved by the Ethics Review Committee of Harbin Medical University (ID: hrbmuecdc20200320) and was consistent with the Helsinki Declaration. Participants in the study all signed informed consent forms.
Methods
Determination of serum iodine and thyroid function indicators
Five milliliters of venous blood (no anticoagulant) was collected from the study subjects and centrifuged at 3000 g after standing at room temperature for 2 hours. Serum was separated and stored at −80°C. Serum iodine concentration was measured using an inductively coupled plasma-MS system (PerkinElmer NexION 350). The levels of fT3, fT4, TSH, TPOAb, and TgAb were determined using chemiluminescent immunoassay (Siemens Healthcare Diagnostics Inc.).
The reference values were 3.1–6.8 pmol/L fT3, 11.5–22.7 pmol/L fT4, 0.27–4.20 μIU/mL TSH, 0–60 U/mL TPOAb, and 0–60 U/mL TgAb.
Determination of urine iodine
From each participant, the fasting single-spot urine sample was collected in the morning (08.00–11.00 hours) in clean labeled plastic tubes and stored at 4°C. The test was completed within two weeks. Urine iodine was measured according to the China Health Standard Method of Determination of Iodine in Urine by As3+-Ce4+ catalytic spectrophotometry (5).
Determination of candidate genes
According to the results of KEGG metabolic pathway enrichment analysis of Illumina Methylation 850K BeadChip in the discovery phase, the methylation of HLA-DPB1, PDCD1LG2, PTPRM, MAG, CDH1, CDH4, and ITGA6 genes related to cell adhesion molecule pathway were closely related to AIT. The methylation status of all the aforementioned genes were evaluated using the MethylTarget™ assay. Subsequently, we mainly considered the distribution and locations of differential methylation sites (DMS) in gene functional regions and the difference in the extent of methylation sites between case and control, and selected HLA-DPB1 and PDCD1LG2 genes for quantitative real-time-polymerase chain reaction (QRT-PCR). The results of the Methylation 850K BeadChip are shown in Supplementary TablesS1–S8.
Determination of methylation
MethylTarget (Genesky Corporation, Shanghai, China) was used to validate the methylation differences of candidate genes. Primer design and optimization was performed by GeneSky Corporation. The primers used are summarized in Table 1. The EZ DNA methylation Kit was used to transform genomic DNA into bisulfite (Zymo, Irvine). Samples were amplified, barcoded, and sequenced (MiSeq; Illumina, Inc., San Diego) using the paired-end sequencing protocol according to the manufacturer's guidelines.
The Sequences of the Primers
Quantitative real-time polymerase chain reaction
RNAiso Plus (Takara, Dalian, China) was used to extract total RNA from whole blood according to the method recommended by manufacturers. Reverse transcription of the sample RNA was performed according to the reverse transcription kit operation manual (Takara). One microliter of diluted cDNA (1:10, vol/vol) were used for real-time PCR, which was performed using a QuantStudio®5 Real-Time PCR instrument (Applied Biosystems). Beta-actin was used as an internal reference and samples were run in triplicate. Primers for real-time PCR were synthesized by Ruibiotech (Beijing, China) and the primer sequence are shown in Table 2. Data were analyzed using the 2−▵▵ CT method and the results are presented as the fold change relative to that of the control group.
Real-Time Fluorescence Quantitative Polymerase Chain Reaction Primer Sequence
Statistical analyses
The methylation levels of CpG site were presented as the percentage of methylated cytosine over total-tested cytosine. All values were expressed as mean ± standard deviation (SD) or percentage (%). Statistical analyses was performed using SPSS 23.0 software (SPSS, Inc., Chicago, IL). Mean ± SD was used for normal distribution data, Student's t-test was used for comparison between two groups, and one-way analysis of variance was used for comparison between multiple groups. For non-normally distributed data, median with interquartile range was used, and the Kruskal–Wallis test was used for comparison between multiple groups. If differences were found between groups, further pairwise comparisons were made accordingly. The correlation between methylation levels of candidate genes and relative mRNA expression were analyzed by Pearson correlation analysis. The graphs were generated using GraphPad Prism Version 9.0 (GraphPad Software, Inc., CA). Two-sided test with p < 0.05 was considered statistically significant.
Results
Demographic information
In this study, 176 patients and 176 healthy controls were included, and there were no statistically significant differences in age, sex, and BMI between the case and control groups. The demographic data for areas with different iodine levels is summarized in Table 3.
Characteristics of Study Subjects from Different Water Iodine Level Areas
Overt hypothyroidism: TSH >4.20 μIU/mL, fT4 < 11.5 pmol/L; subclinical hypothyroidism:TSH >4.20 μIU/mL, fT4 within the normal range. Hypothyroid including cases of overt hypothyroidism and subclinical hypothyroidism.
The case group compared with the control group in each area, p < 0.05.
IA areas, iodine-adequate areas; IE areas, iodine-excess areas; IF areas, iodine-fortification areas; SIC, serum iodine concentration; TSH, thyrotropin; UIC, urinary iodine concentration.
Analysis of methylation levels in CpG sites of candidate genes
Samples from all participants were used for the MethylTarget assay validation. Six target regions of the candidate genes were sequenced, including HLA-DPB1_1, HLA-DPB1_2, HLA-DPB1_3, HLA-DPB1_4, HLA-DPB1_5, and PDCD1LG2_1. To better understand the DNA methylation of HLA-DPB1 and PDCD1LG2 genes related to the cell adhesion molecule pathway, we evaluated the methylation level of CpG sites in each target region. The 57 CpG sites were subjected to differential methylation analyses, and the results revealed that only 4 of the 54 CpG sites in HLA-DPB1 gene had significantly lower methylation levels in AIT patients compared with controls (all p < 0.05), and there was no significant difference in the methylation of CpG sites for the PDCD1LG2 gene between the two groups (all p > 0.05) (Table 4). The methylation levels of CpG sites in the target regions of each candidate gene are shown in Figure 1.
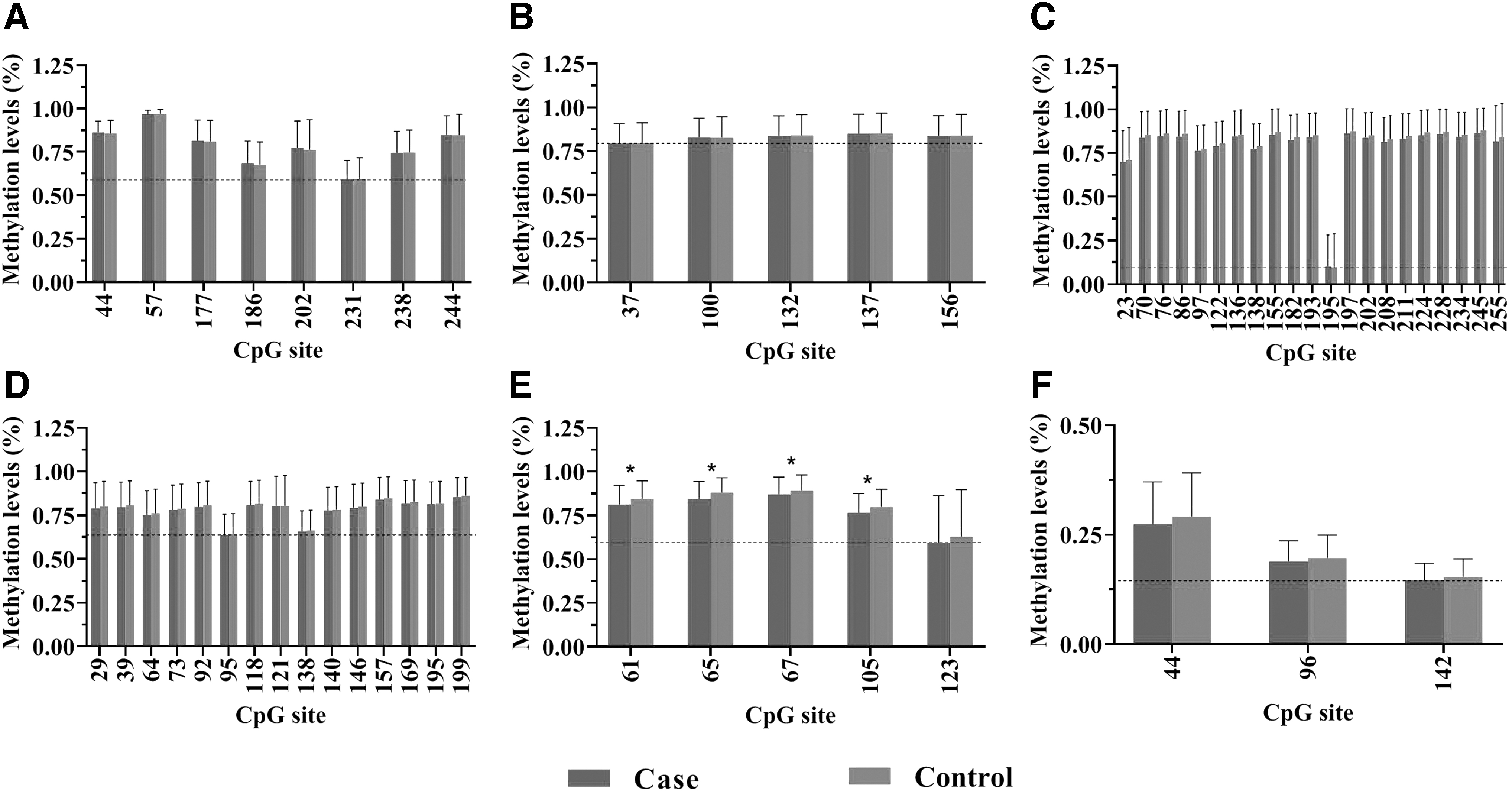
Methylation level differences of CpG sites in the corresponding target regions of candidate genes between case and control groups; *The case group compared with the control group in each CpG site, p < 0.05. (
Validations of Differential Methylation Sites and the Relative mRNA Expressions from Two Differentially Methylated Genes
Positive sites by methylation of 850K BeadChip.
The case group compared with the control group, p < 0.05.
“−” downregulated; “+” upregulated; Chr, chromosome; FC, fold change; Meth. Diff, the methylation level of case − the methylation level of control (the positive number indicates hypermethylation in the case group compared with the control group, while the negative number indicates hypomethylation in the case group compared with the control group); QRT-PCR, quantitative real-time-polymerase chain reaction.
Analysis of the difference of methylation levels of candidate gene CpG sites in populations from areas with different water iodine levels
The difference in the average methylation level of candidate genes in CpG sites between the case group and the control group in areas with different water iodine levels is shown in Table 5. The results showed that in the case group and the control group of IF areas, there were significant differences in methylation levels of three target regions of two genes covering seven CpG sites, including HLA-DPB1 (chr6: 33048602, chr6: 33048875, chr6: 33048879, chr6: 33048881, chr6: 33048919, and chr: 33048937; all p < 0.05) and PDCD1LG2 (chr9: 5510544; p = 0.017). The DMS of HLA-DPB1 and PDCD1LG2 genes showed lower methylation level in the case group than in the control group. There was no significant difference in the methylation level of other CpG sites (p > 0.05). For IA areas, the methylation levels of HLA-DPB1 and PDCD1LG2 genes in the case group were lower than that in the control group, but there was no significant difference in the methylation levels of the corresponding CpG sites of these candidate genes. In the IE areas, only two target regions of HLA-DPB1 gene covered three CpG sites with significant differences in methylation levels (chr6: 33048077, chr6: 33048735, and chr6: 33048809; all p < 0.05), and the methylation levels of each were hypermethylation in the case group compared with the control group, while the methylation levels of other CpG site showed no statistically significant differences (all p > 0.05).
CpG Methylation Levels of Candidate Genes in Areas with Different Water Iodine Levels Between Case and Control Groups
Positive sites by methylation of 850K BeadChip.
The case group compared with the control group, p < 0.05.
Correlation analysis between DNA methylation and relative mRNA expression of HLA-DPB1 and PDCD1LG2 genes in AIT
The target region HLA-DPB1_3, of the HLA-DPB1 gene, was negatively correlated with the relative mRNA expression level of HLA-DPB1 gene (r = 0.107, p = 0.047). The average methylation level of the target region PDCD1LG2_1, of the PDCD1LG2 gene, was negatively correlated with the relative mRNA expression level of PDCD1LG2 gene (r = 0.135, p = 0.011). There was no significant difference between the target region of other genes and the relative mRNA expression level of the specific gene (Table 6).
Correlation Between DNA Methylation and Relative mRNA Expression in HLA-DPB1 and PDCD1LG2 Genes
r, Pearson correlation coefficient.
Correlation analysis, p < 0.05.
The relationship between DNA methylation of HLA-DPB1 and PDCD1LG2 genes and their relative mRNA expression levels in the areas with different water iodine levels showed that the average methylation levels of the target region HLA-DPB1_1, HLA-DPB1_3, and HLA-DPB1_5 of HLA-DPB1 gene in IF areas were negatively correlated with the relative mRNA expression level of HLA-DPB1 gene (r = −0.156, p = 0.040; r = −0.167, p = 0.027; r = −0.178, p = 0.017). For IA areas, the average methylation level of the target region PDCD1LG2_1 of PDCD1LG2 gene was negatively correlated with the relative mRNA level of PDCD1LG2 (r = −0.221, p = 0.048). There was no significant correlation between other target regions and relative mRNA expression levels (Table 6).
Discussion
HLA-DPB1 is a member of the immunoglobulin superfamily of cell adhesion molecules and belongs to the class of HLAII molecules. The gene has the function of antigen presentation, which is directly involved in the activation of T cells, as well as in the regulation of the adaptive immune response (4). It has been suggested that the β chain of HLA-DP, a class of HLAII molecules, may be involved in the molecular pathogenesis of early-onset AIT (6). However, it is unknown whether the occurrence of AIT is related to DNA methylation of HLA-DPB1 gene. In this study, we found that the CpG site 61 (chr6:33048875) of the target region HLA-DPB1_5 was consistent with the previous methylation BeadChip results, that is, the case group showed a lower methylation state than the control group. This difference was present for IF areas. The target region and CpG site of HLA-DPB1 gene detected in this study were located in the gene body region. Some studies have suggested that the transcriptional level of genes with hypermethylation level in the gene body region is in the middle, while decreasing the DNA methylation level in the gene body region can activate gene expression (7). Therefore, we hypothesize that the hypomethylation state of the target region activated the expression of this gene, thus promoting the activation of T cells, thereby initiating or aggravating the thyroid autoimmune response. These results suggest that the low methylation levels of the CpG site in the target region of HLA-DPB1 gene may play an important role in the pathogenesis of AIT.
PDCD1LG2 gene, located on chromosome 9, is a member of the cell adhesion molecule immunoglobulin superfamily and one of the ligands of programmed cell death 1 (PDCD1). PDCD1LG2 can induce immune tolerance under physiological and pathological conditions, and in thyroid cancer-related studies, the level of DNA methylation of the gene is negatively correlated with immune cell infiltration (8). However, the relationship between the gene and AIT is unknown. Our results showed that methylation levels in the validated target region PDCD1LG2_1 and the target region CpG site 44 (chr9: 5510596) did not differ significantly between the case and control groups. However, when we analyzed the methylation differences among people with different water iodine levels, we found that the methylation levels of target region PDCD1LG2_1 and CpG site 96 (chr9: 5510544) were significantly different between cases and controls in IF areas, but the difference was not significant in IA and IE areas. Although the iodine nutrition level of the population in the IF areas is close to that in the IA areas, the IF areas used to be in a state of iodine deficiency for a long time, and the iodine nutrition level of the population in this area has been improved after iodine fortification in recent years. Many previous studies have found that the incidence of iodine-induced hyperthyroidism will increase in the years after iodine fortification in iodine-deficient areas, and some studies have found that the incidence of AIT diseases increases after iodine fortification in iodine-deficient areas (9,10). In this study, the long-term iodine deficiency state in the past and the adaptive changes of the body after iodine fortification may induce epigenetic changes in some people susceptible to thyroid diseases in IF areas. As a result, some changes may have taken place in the methylation level of some genes, but this needs to be further studied.
It is well known that DNA methylation plays an important role in gene transcriptional regulation. In this study, we found that multiple target regions of HLA-DPB1 gene and PDCD1LG2 gene were negatively correlated with their relative mRNA expression levels. Although the correlation was weak, it may impact gene expression levels.
At present, the role of genomic methylation in gene regulation is not completely clear. One hypothesis has been proposed that intragenic methylation can affect gene expression levels by reducing transcriptional elongation (11,12). In this case, the level of methylation should be negatively correlated with the level of gene expression (i.e., hypermethylation and low expression of mRNA). Similar to this view, the methylation sites and regions of the HLA-DPB1 gene validated in this study were located in the gene body region, and we found that there was a negative correlation between partial methylation of the HLA-DPB1 gene and gene expression in whole blood, which is consistent with the traditional concept of DNA methylation as an inactive chromatin marker (13). Some researchers believe that the high expression of HLAII genes may turn itself into antigen-presenting cells and induce autoimmune response (14).
Multiple studies have shown that DNA methylation downstream of the transcription start site was more closely related to gene transcription inhibition than upstream of the transcription start site (i.e., the promoter region) (15,16). In our study, the methylation site and region of PDCD1LG2 gene we validated were located in the first exon region of the genome (downstream from the transcription start site −109 bp). At the same time, we also found that there was a negative correlation between PDCD1LG2 gene region methylation and gene expression in whole blood (i.e., hypermethylation and low mRNA expression). This is similar to the findings of Sakamoto et al. (13). They showed that in gastric cancer mouse model, the low methylation in the CpG-rich region in the first exon of Twist1 gene was closely related to the activation of Twist1 gene in mouse gastric cancer cells (14). These results suggest that the hypomethylation of PDCD1LG2 gene may indicate the dynamic change of methylation pattern and affect the gene expression level during the development of AIT. However, the role of DNA methylation in this gene and its relationship with AIT need to be further investigated. Based on our results, we hypothesize that the hypomethylation of PDCD1LG2 gene in AIT patients may lead to an increase in mRNA expression, thus affecting the development of AIT.
This study has several strengths and limitations. First, in this study, we analyzed the differences in DNA methylation patterns of candidate genes in areas with different water iodine levels, which have rarely been reported in previous studies. Second, we integrated the DNA methylation level of candidate genes with transcriptome data, which plays an important role in identifying candidate gene DNA methylation in the transcriptional regulation of related genes. This study also had some limitations. In this study, we only studied the relationship between candidate gene DNA methylation and AIT, but did not study the molecular functional mechanism.
In conclusion, our study shows a change in the methylation status of genomic DNA in patients with AIT. There were hypomethylated regions and sites in the gene body region of HLA-DPB1 and PDCD1LG2 genes, and we found that the relative mRNA of these two genes was upregulated in AIT cases and AIT cases with different water iodine levels. These results suggest that the HLA-DPB1 and PDCD1LG2 genes related to cell adhesion molecule pathway may be involved in the development of AIT.
Footnotes
Acknowledgments
The authors are grateful for the assistance provided by the Institute for Prevention and Treatment of Endemic Disease of Shandong Province for collecting epidemiological data and samples, and contributions and support from all participants.
Authors' Contributions
The contribution of each author is as follows: H.S. designed the study; H.S., S.W., L.L., B.R., M.Q., H.W., W.J., and X.W. conducted the research. S.W. analyzed the data. S.W. and H.S. drafted the article. All authors revised the report and approved the final version before submission.
Author Disclosure Statement
The authors hereby confirm that no part of this article has been published or is under consideration for publication elsewhere. The authors have no potential conflicts of interest to declare.
Funding Information
This study was supported by the National Natural Science Foundation of China (Grant No. 81872561) and Scientific Research and Practice Innovation Fund for Postgraduate of Harbin Medical University (Grant No. YJSKYCX2019-16HYD).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8