
Abstract
Background:
A recent genome-wide association study identified the SLC17A4 locus associated with circulating free thyroxine (T4) concentrations. Human SLC17A4, being widely expressed in the gastrointestinal tract, was characterized as a novel triiodothyronine (T3) and T4 transporter. However, apart from the cellular uptake of T3 and T4, transporter characteristics are currently unknown. In this study, we delineated basic transporter characteristics of this novel thyroid hormone (TH) transporter.
Methods:
We performed a broad range of well-established TH transport studies in COS-1 cells transiently overexpressing SLC17A4. We studied cellular TH uptake in various incubation buffers, TH efflux, and the inhibitory effects of different TH metabolites and known inhibitors of other TH transporters on SLC17A4-mediated TH transport. Finally, we determined the effect of tunicamycin, a pharmacological inhibitor of N-linked glycosylation, and targeted mutations in Asn residues on SLC17A4 function.
Results:
SLC17A4 induced the cellular uptake of T3 and T4 by ∼4 times, and of reverse (r)T3 by 1.5 times over control cells. The uptake of T4 by SLC17A4 was Na+ and Cl− independent, stimulated by low extracellular pH, and reduced by various iodothyronines and metabolites thereof, particularly those that contain at least three iodine moieties irrespective of the presence of modification at the alanine side chain. None of the classical TH transporter inhibitors studied attenuated SLC17A4-mediated TH transport. SLC17A4 also facilitates the efflux of T3 and T4, and to a lesser extent of 3,3′-diiodothyronine (T2). Immunoblot studies on lysates of transfected cells cultured in absence or presence of tunicamycin indicated that SLC17A4 is subject to N-linked glycosylation. Complementary mutational studies identified Asn66, Asn75, and Asn90, which are located in extracellular loop 1, as primary targets.
Conclusions:
Our studies show that SLC17A4 facilitates the transport of T3 and T4, and less efficiently rT3 and 3,3′-T2. Further studies should reveal the physiological role of SLC17A4 in TH regulation.
Introduction
Transporter proteins are a prerequisite for transfer of thyroid hormones (THs) across the plasma membrane (1,2). So far, several members of the organic anion transporting polipeptide (OATP) family, L-type amino acid transporter (LAT) 1 and LAT2, sodium taurocholate cotransporting polypeptide (NTCP), monocarboxylate transporter (MCT) 8, and MCT10, have been identified as TH transporters [reviewed in Refs. (2 –4)]. Each of these transporters has its own substrate specificity and tissue distribution; the majority of transporters accept a broad variety of other substrates. The most specific TH transporter identified to date is MCT8 that facilitates cellular transport of triiodothyronine (T3) and thyroxine (T4), and to a lesser extent reverse (r)T3 and 3,3′-diiodothyronine (T2) (5,6).
Mutations in SLC16A2, encoding MCT8, cause MCT8 deficiency, a disorder with neurodevelopmental and metabolic features (7,8). Mutations in the brain-specific T4 transporter OATP1C1 have been recently linked to a neurodegenerative phenotype (9). These studies have substantiated the physiological relevance of TH transporters.
In a recent genome-wide association study, we demonstrated that genetic variation in SLC17A4 was associated with serum-free T4 concentrations (10). Functional studies indicated that SLC17A4 facilitates the uptake of T3 and T4 as efficiently as MCT8 (10). The SLC17 family consists of 9 structurally related proteins, of which the first 4 members (SLC17A1–4) had been initially classified as Na+-dependent inorganic phosphate transporters (11).
Over the years, it turned out that SLC17A1–3 accept a broad range of organic anions (11). However, SLC17A4 has long been designated as an orphan transporter (11). The few available studies indicated strong SLC17A4 mRNA expression in liver, kidney, small intestine, colon, and pancreas in human and rat (12,13). Its transport characteristics have only been studied in proteoliposomal systems, in which it induced the accumulation of uric acid (12).
Although SLC17A4 mediates the cellular uptake of T3 and T4, it is yet unknown whether SLC17A4 also facilitates TH efflux. Likewise, it is currently unknown whether SLC17A4 also accepts other iodothyronines and metabolites thereof as a substrate. Finally, it is important to address whether SLC17A4 is sensitive toward frequently applied pharmacological inhibitors of other TH transporters such as silychristin and 2-aminobicyclo[2.2.1]heptane-2-carboxylic acid (BCH). However, knowing all these key features is mandatory to position SLC17A4 among the currently known TH transporters. Moreover, such information will inform the assessment of the physiological function of SLC17A4 in in vivo models.
To close this gap in current knowledge, we here delineated the basic functional characteristics of the human SLC17A4 protein.
Materials and Methods
Expression constructs, cell culture, and transfection
A pCMV6-Entry_human SLC17A4 expression vector containing a C-terminal Myc and Flag tag was obtained from OriGene Technologies (Rockville, USA). The cloning of human μ-crystallin (pSG5-hCRYM) has been previously described (14). COS-1 African green monkey kidney cells (CVCL_0223) were obtained from ECACC (Sigma–Aldrich, Zwijndrecht, The Netherlands) and cultured and transfected under standard conditions (15,16). Technical details are provided in the Supplementary Data.
Functional studies
TH uptake, efflux, and metabolism assays, as well as cell surface biotinylation, immunoblotting, and immunocytochemistry studies were essentially performed as recently described (15,16). Full technical details are available in the Supplementary Data and Supplementary Table 1. To facilitate comparison with other TH transporters that we have previously characterized [e.g., Refs. (6,14,17,18)], we performed all studies in the same COS-1 African green monkey kidney cell line. Alternative cell lines available in our laboratory that yield sufficient transfection potency did not show endogenous SLC17A4 mRNA expression (Supplementary Fig. S1A, B), and were, therefore, not deemed to provide a more suitable model for the natural cellular context of SLC17A4 than COS-1 cells.
SLC17A4 homology modeling
An SLC17A4 homology model was constructed similarly as has recently been done for MCT8 using YASARA Structure Software (16,19,20).
Statistics
All statistical analyses were carried out using GraphPad Prism version 6 (GraphPad Software, San Diego, CA, USA). The applied statistical tests and levels of significance are mentioned in the legend of the corresponding figure.
Results
Upon transfection of COS-1 cells with increasing amounts of SLC17A4 plasmid, a dose-dependent increase of T4 uptake was observed with an apparent optimum at 250 ng (Fig. 1A). In line with previous studies, the intracellular accumulation of T4 was enhanced in the presence of μ-crystallin (CRYM), with a plateau starting at an SLC17A4 plasmid dose of ∼50 ng (Fig. 1A). Therefore, we selected the 250 and 125 ng dose for further experiments in absence and presence of CRYM, respectively. Similarly, a dose-dependent increase in SLC17A4 protein expression levels was observed, with immunoreactive bands at ∼47 and ∼55 kDa, which are close to the predicted size of the SLC17A4 monomer (∼54 kDa), and at ∼90–100 kDa, which may correspond with SLC17A4 (hetero-)dimer (Fig. 1B).

(
To study the Na+ dependence of SLC17A4-mediated T4 transport in transfected COS-1 cells, T4 uptake was measured in NaCl uptake buffer and in Na+-free uptake buffer in which NaCl was replaced by an equimolar amount of choline chloride. Elimination of extracellular Na+ did not affect T4 uptake by SLC17A4, indicating that the transport of T4 is Na+ independent (Fig. 2A). T4 transport was also not sensitive to alterations in membrane potential because replacing extracellular Na+ with K+ (which depolarizes the cell membrane) did not change T4 uptake rate (Fig. 2B).
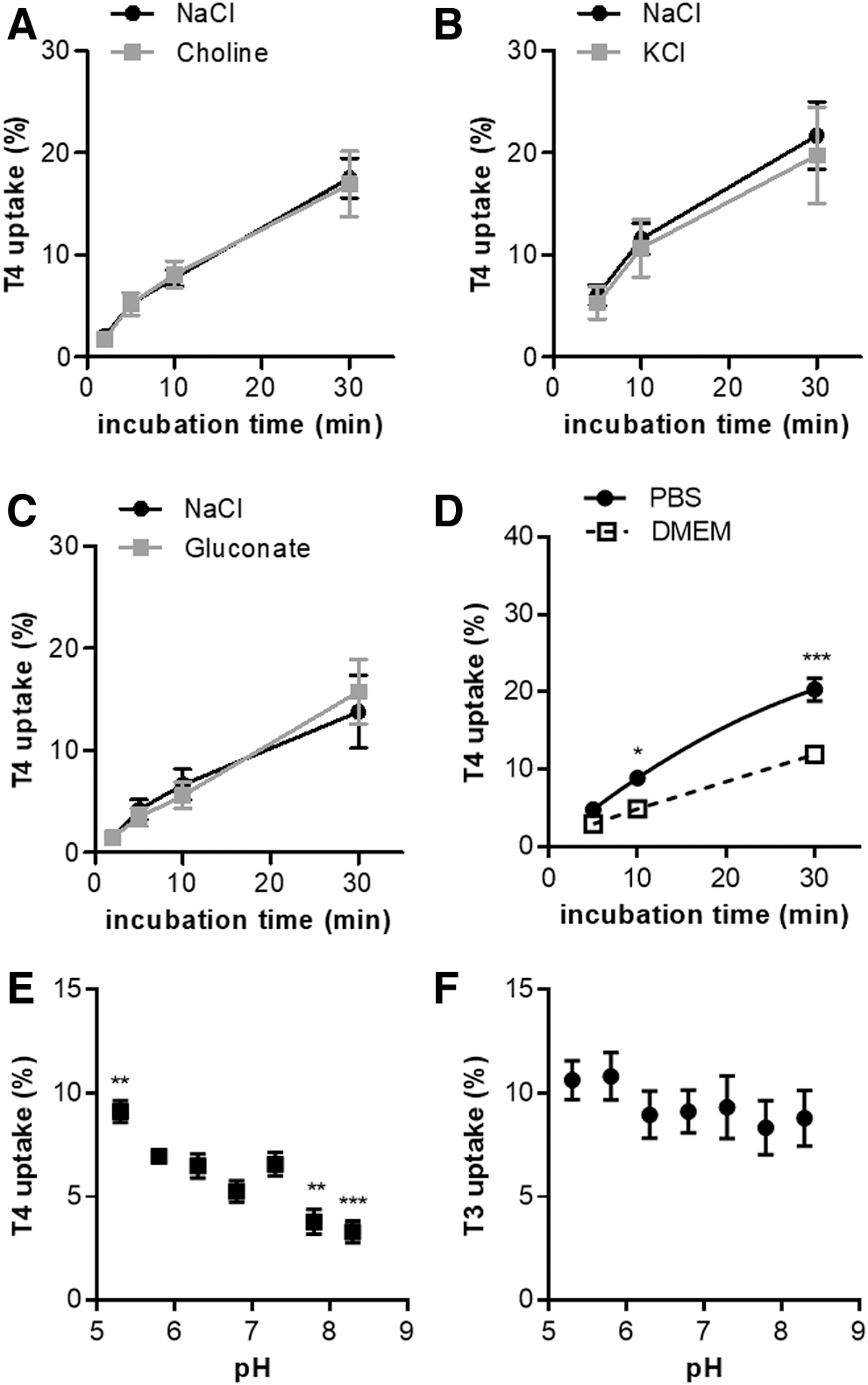
(
Previous studies in proteoliposomes showed that Cl− is indispensable for the transport of at least some substrates by various members of the SLC17 family (12,21,22). However, creating an outward gradient of Cl− by replacing the extracellular Cl− by gluconate did not change the T4 uptake rate (Fig. 2C), indicating that SLC17A4 is not a Cl−/T4 exchanger. Also in Dulbecco's modified Eagle's medium (DMEM) incubation, in which vitamins and nutrients are present at (near-) physiological concentrations, SLC17A4 induced T4 uptake, although the observed accumulation levels after 10 and 30 minutes incubation were lower than in Dulbecco's phosphate buffered saline (Fig. 2D).
Similar results were obtained in studies performed with T3 as a substrate (Supplementary Fig. S2A–D). SLC17A4-mediated T4 uptake was pH sensitive, with T4 uptake levels at pH 5.3 significantly exceeding those observed at neutral pH (7.3). By contrast, T4 uptake was reduced at (slightly) basic pH (7.8 and 8.3) (Fig. 2E). A similar pattern was observed for SLC17A4-mediated T3 uptake, although the differences between the conditions did not reach statistical significance (Fig. 2F). The uptake of T4, but not T3, was also enhanced at pH <6.3 in empty vector control cells (data not shown).
Next, we delineated the substrate specificity of SLC17A4. In the absence of CRYM, SLC17A4 induced T3 and T4 uptake by ∼2 times and that of rT3 by 1.8 times over control cells, whereas 3,3′-T2 uptake was not induced (Fig. 3A). In the presence of CRYM, a similar substrate preference was observed, with the induction of T3 and T4 uptake (∼4 times) exceeding the induction of rT3 (1.8 times) and 3,3′-T2 (1.3 times) accumulation (Fig. 3B). SLC17A4 showed a higher specificity toward T3 and T4 compared with MCT8, which also substantially induced the cellular uptake of 3,3′-T2 (Supplementary Fig. S3). Coexpression of SLC17A4 and type 3 deiodinase (D3) greatly increased the intracellular deiodination of T3 (Fig. 3C) and T4 (Fig. 3D) compared with cells transfected with SLC17A4 or D3 alone.
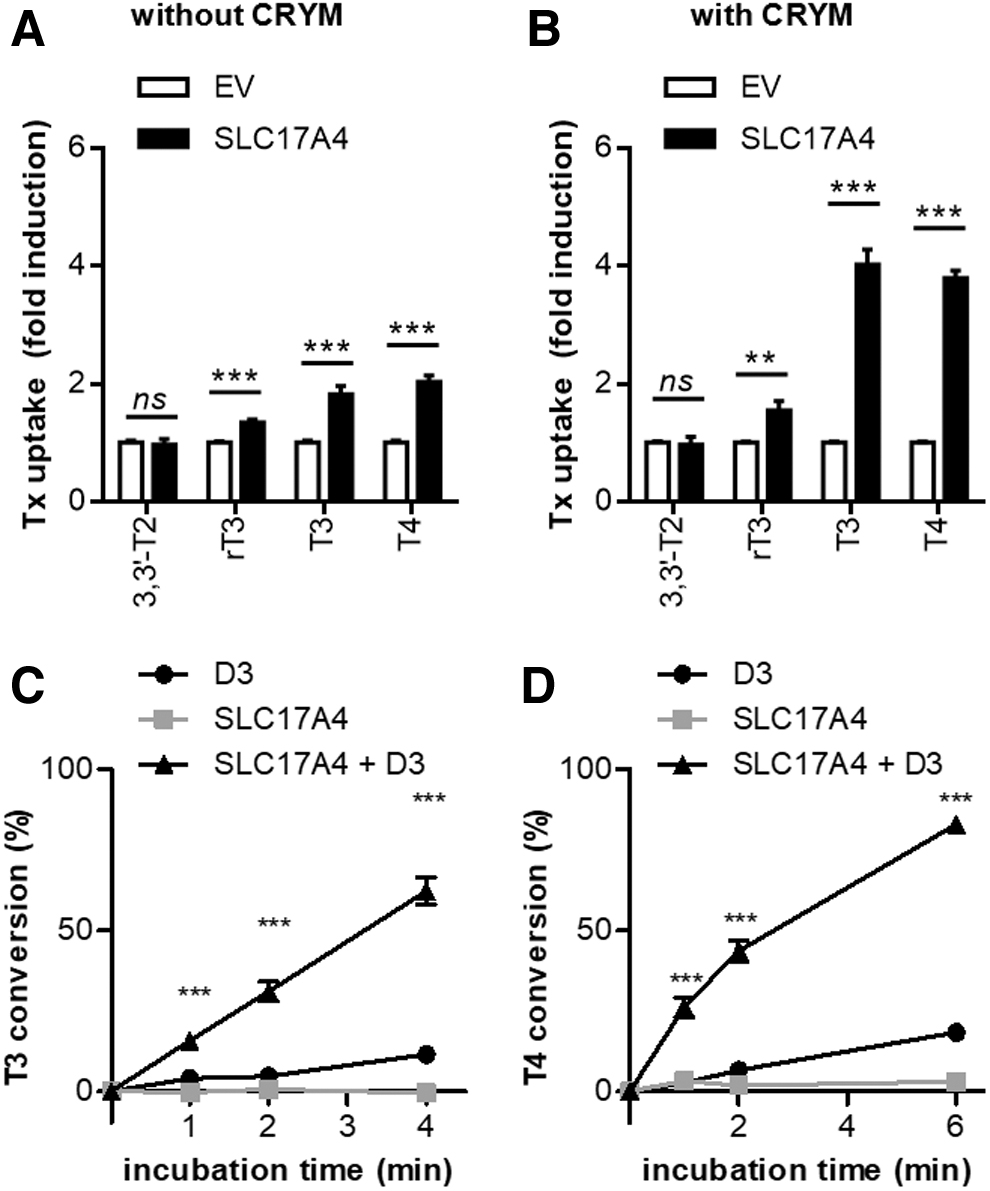
Substrate specificity of SLC17A4. (
Since the uptake levels in the presence of CRYM exceeded those observed in the absence of CRYM, we next studied whether SLC17A4 also facilitates the cellular efflux of iodothyronines. After preloading transfected COS-1 cells with radiolabeled iodothyronines for two hours, the amount of intracellular 3,3′-T2, rT3, T3, and T4 was measured after different incubation times in efflux medium. The decline in cellular 3,3′-T2 and rT3 content did not differ between COS-1 cells expressing SLC17A4 versus control cells, except for a minimal difference in cellular 3,3′-T2 levels after five minutes in favor of SLC17A4 expressing cells (Fig. 4A, B). In contrast, T3 and T4 efflux was markedly faster from cells transfected with SLC17A4 than from control cells (Fig. 4C, D).

(
The substrate preference of SLC17A4 was addressed further by investigating the uptake of [125I]-T3 in the presence or absence of various structurally related compounds. The first group of compounds constituted various iodothyronines that differ from T3 in number and position of the iodine moieties. At 1 μM concentrations, T3 uptake was slightly, but significantly, reduced by 3,3′-T2, 3,5-T2, and 3′,5′-T2, but not T0. By contrast, T3 uptake was largely diminished in the presence of 1 μM T3 or T4, which is in line with previously reported IC50 values of <1 μM for these substrates (10).
At a higher concentration (10 μM), 3,3′-T2, 3,5-T2, and to a lesser extent 3′,5′-T2 also reduced SLC17A4-mediated T3 uptake, whereas T0 had still no effect (Fig. 5A). T3 uptake was (near-) completely blocked in the presence of 10 μM rT3, T3, and T4 (Fig. 5A).
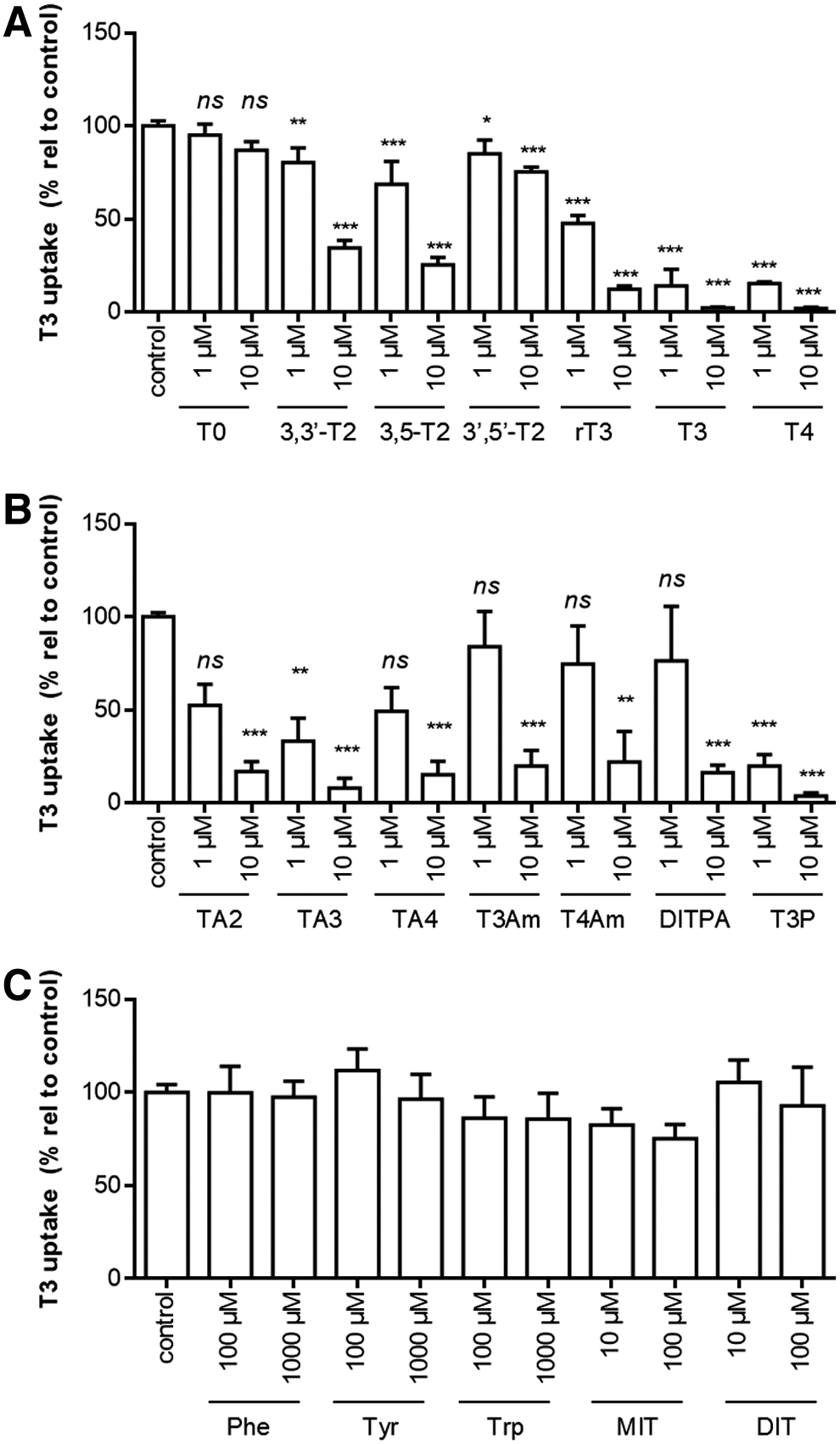
(
The second group of compounds constituted various alanine side-chain metabolites, including iodothyroacetic (TAx) and iodothyroproprionic (TPx) acid derivatives as well as thyronamines (TxAm). In particular, TA3 and T3P reduced SLC17A4-mediated T3 uptake at 1 μM concentrations, whereas all compounds largely diminished T3 uptake at 10 μM concentrations (Fig. 5B).
The third group of compounds constituted the aromatic amino acids and the iodotyrosines monoiodotyrosine (MIT) and di-iodotyrosine (DIT), which differ from T3 due to the absence of the phenolic outer ring and/or the inner ring iodine moieties. At 100 or 1000 μM concentrations, these compounds did not affect SLC17A4-mediated T3 uptake (Fig. 5C). These findings were confirmed by similar experiments conducted with [125I]-T4 in the absence (data not shown) and presence (Supplementary Fig. S4A, B) of CRYM.
To study whether compounds that potently inhibited SLC17A4-mediated TH uptake are also genuine substrates for SLC17A4, we assessed the uptake of [125I]-TA3 and [125I]-TA4 in COS-1 cells transfected with SLC17A4 or empty vector control. Indeed, the uptake of [125I]-TA3 was slightly, but significantly, induced by SLC17A4, whereas [125I]-TA4 uptake was not enhanced (Fig. 6A, B). As SLC17A4 is expressed along the gastrointestinal tract, we also studied the uptake of the sulfo-conjugates [125I]-TS3 and [125I]-TS4. In contrast to OATP1B1, known as a potent transporter of both substrates (18), SLC17A4 did not induce the uptake of T3S (Fig. 6C), whereas a small, but significant, induction of T4S uptake was observed (Fig. 6D).
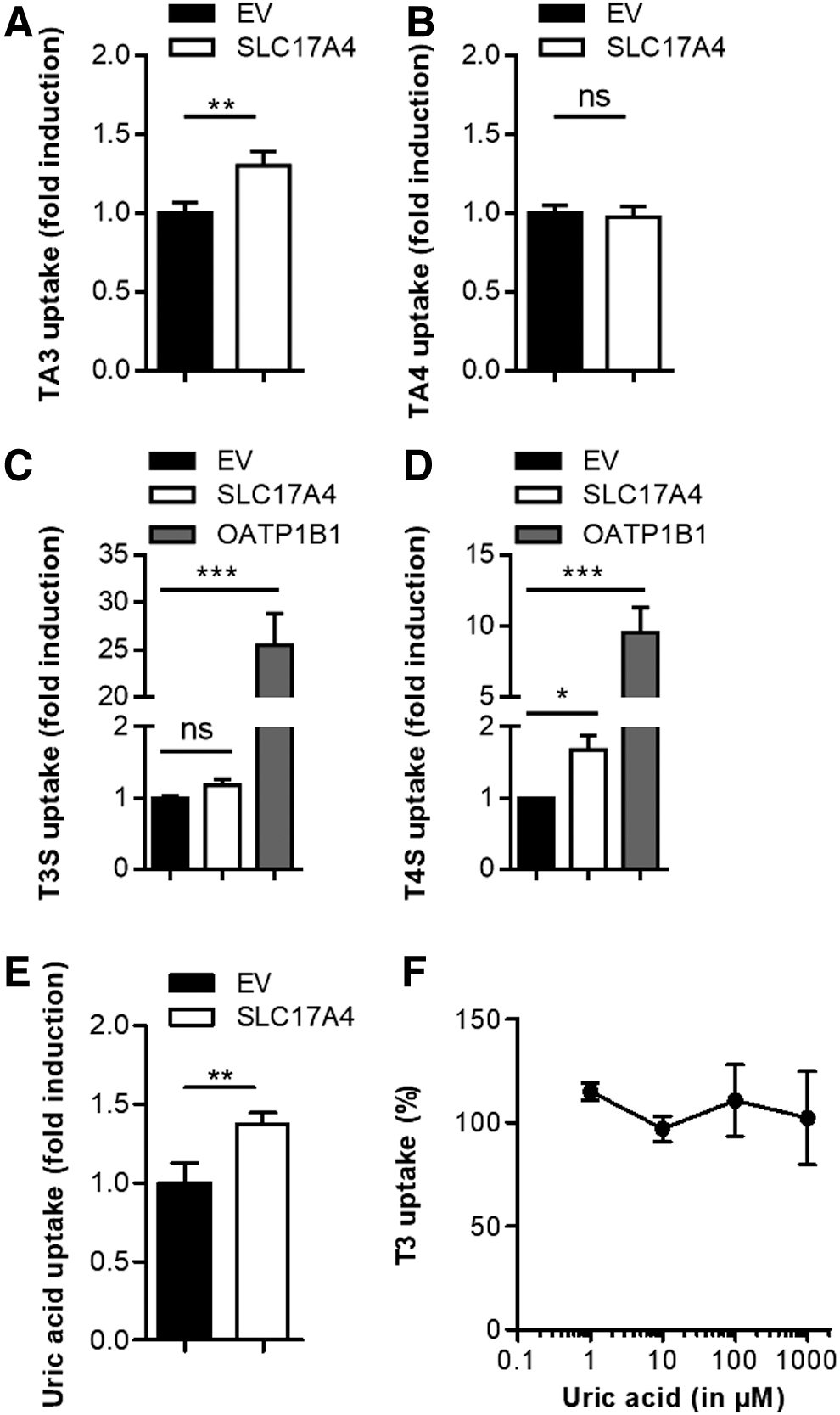
(
Although less pronounced than in proteoliposomes (12), uric acid uptake in SLC17A4-transfected COS-1 cells was increased by ∼1.4 times over control cells in which uric acid uptake rate varied between 3 and 15 pmol/min (Fig. 6E). However, even at a concentration of 1000 μM, which is well above the reported reference ranges in serum [e.g., Ref. (23)], uric acid had no effect on SLC17A4-mediated T3 uptake (Fig. 6F).
We also studied the impact of well-established inhibitors of other TH transporters on SLC17A4-mediated T3 uptake, including naringin (OATP1A2, and possibly other OATPs), BCH (LATs), sulfobromophthalein (BSP) and verapamil (multiple transporters), silychristin (MCT8), and estrone sulfate (E3S; substrate for NTCP and OATPs). However, none of these compounds reduced SLC17A4-mediated T3 (Fig. 7) or T4 (Supplementary Fig. S4C) uptake when applied at concentrations known to reduce the function of other TH transporters.
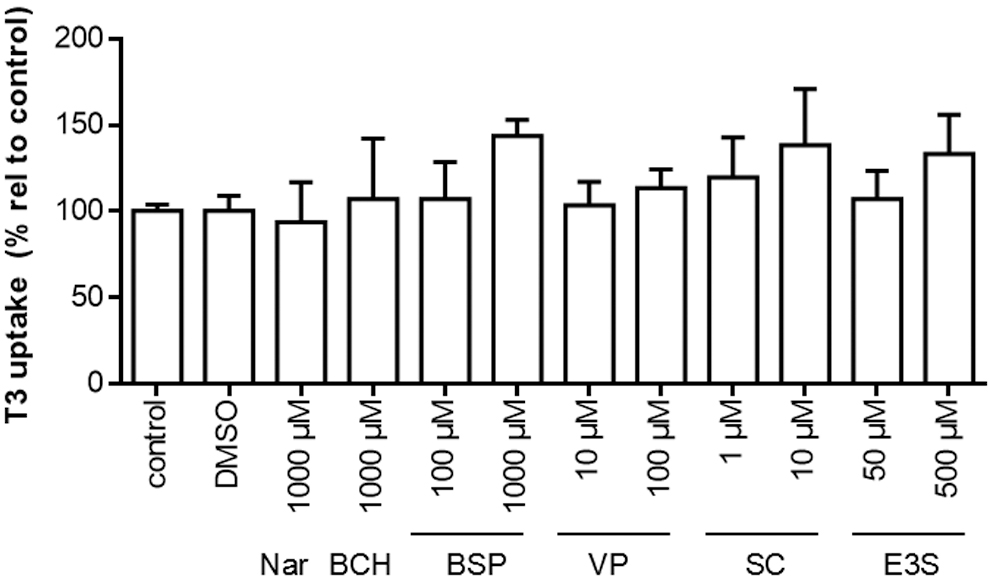
T3 uptake in the presence or absence of the indicated known inhibitors of other well-established TH transporters after 30 minutes incubation at 37°C. All uptake levels are corrected for background T3 uptake in EV-transfected control cells incubated in the presence of the same inhibitor. T3 uptake levels are displayed relative to the amount of intracellular [125I]-T3 in cells incubated in the absence of any competitors (100%) and presented as means ± SEM (n = 3). All experiments were carried out in COS-1 cells cotransfected with CRYM. A one-way ANOVA was carried out followed by Dunnett's post-tests to assess for significant differences between the T3 uptake levels in the absence or presence of the tested inhibitors. BCH, 2-aminobicyclo[2.2.1]heptane-2-carboxylic acid; BSP, sulfobromophthalein; E3S, estrone sulfate; nar, naringin; SC, silychristin; VP, verapamil.
Finally, we studied whether SLC17A4 is subject to glycosylation. In contrast to transfected cells cultured in normal culturing medium, which showed immune-reactive bands at 47 and 54 kDa, cells cultured in the presence of the N-linked glycosylation blocker tunicamycin only showed an immunoreactive band at 47 kDa (Fig. 8A), suggesting that the 54 kDa band represents an N-linked glycosylated form of SLC17A4. Parallel uptake studies showed a strong reduction of SLC17A4-mediated T3 and T4 uptake in the presence of tunicamycin (Fig. 8B and Supplementary Fig. S5A).

(
N-linked glycosylation prediction tools, based on the presence of Asn-Xaa-Ser/Thr sequons and topology predictions, identified Asn47, Asn56, Asn66, Asn75, and Asn90 as putative target sites for N-linked glycosylation. To further refine these predictions, we generated an SLC17A4 protein homology model based on the crystal structure of the
The TMDs are interconnected by intra- and extracellular loops, of which the first extracellular loop (ECL1) and third intracellular loop (ICL3) are relatively large. Using the SLC17A4 homology model, we found that only Asn66, Asn75, and Asn90 were located at accessible positions in ECL1, whereas Asn47 and Asn56 were buried within TMD1 (Fig. 8C). In contrast to wild-type SLC17A4, a triple mutant in which these three Asn residues were substituted by an Ala only showed a prominent band at ∼47 kDa (Fig. 8D), corresponding to the band observed after treatment with tunicamycin (Fig. 8A). Concordantly, the triple mutant exhibited a functional reduction of ∼70% compared with wild-type SLC17A4 (Fig. 8E and Supplementary Fig. S5B).
Discussion
In this study, we detailed the functional characteristics of the novel TH transporter SLC17A4. We demonstrated that SLC17A4 primarily mediates the cellular uptake and efflux of T3 and T4. SLC17A4-mediated TH uptake was diminished by a wide range of iodothyronines and alternate alanine side-chain metabolites, but not by iodotyrosines, aromatic amino acids, or classical TH transporter inhibitors. Finally, we showed that SLC17A4 is subject to N-linked glycosylation, which likely targets Asn residues in the ECL1. These findings will help positioning SLC17A4 among the well-established TH transporters.
Compared with MCT8, generally considered the most specific iodothyronine transporter identified to date (5,26), SLC17A4 yields an even higher specificity toward T3 and T4. Like MCT8 (24), SLC17A4 increases the intracellular availability of T3 and T4 for deiodination that confirms that SLC17A4 controls intracellular TH homeostasis by regulating the transport of TH across the cell membrane. Moreover, the apparent Km values of SLC17A4 for T3 (0.41 ± 0.14 μM) and T4 (0.18 ± 0.17 μM) transport (10) are ∼10–20 times lower than those reported for MCT8 as well as most other known TH transporters (27), further supporting a putative physiological role of SLC17A4 in cellular TH homeostasis.
Cis-inhibition studies suggested that SLC17A4 may also recognize other iodothyronines and alanine side-chain metabolites thereof, including the TPx acid and TAx acid derivatives as well as the TxAm. The most effective cis-inhibitory compounds contained two phenolic rings with two inner-ring iodine and at least one outer-ring iodine moiety, whereas the presence of any modifications to the alanine side chain made no apparent differences and might not be critical for initial substrate recognition. By contrast, direct uptake studies indicated that TA3 is a less suitable substrate for SLC17A4 than T3, whereas TA4 was not directly transported by SLC17A4 at all.
Therefore, we speculate that the composition of the alanine side chain is critical to induce conformational changes that lead to the actual transport of the substrate. In light of these findings, it should be emphasized that other compounds that exhibited pronounced cis-inhibitory effects in our studies are not necessarily genuine substrates for SLC17A4. None of the classical TH transporter inhibitors reduced SLC17A4-mediated TH uptake. Therefore, there is no need to reconsider their specificities toward the well-established TH transporter systems.
As SLC17A4 belongs to a different transporter family than previously identified TH transporters (2), it apparently lacks the moieties targeted by these classical inhibitors. As many of the tested inhibitors also block transporters that do not transport iodothyronines, some of which even belong to different protein families, we speculate that these inhibitors target moieties other than the TH recognition site(s) within the substrate binding pocket. SLC17A4 has been previously shown to exhibit membrane potential (Δψ) and Cl−-dependent transport of urate and p-aminohippuric acid (PAH) in proteoliposomes (12). However, our functional studies in transiently transfected mammalian COS-1 cells indicated that SLC17A4 does not require Cl− to transport T4. Moreover, T4 transport was not attenuated by changes in membrane potential secondary to high extracellular K+ concentrations. T4 transport was Na+ independent and was stimulated at low extracellular pH. It is yet unclear whether the transport of TH is proton coupled, given the pronounced residual T4 uptake at basic pH and the limited impact of extracellular pH on T3 transport.
Alternatively, changes in extracellular pH may also affect the protonation state of the substrate and/or amino acid side chains at the substrate binding pocket of the transporter. The lower apparent pKa of the phenolic hydroxyl group of T4 (6.7) versus T3 (8.5) may explain why the uptake of T3 appears to be less affected across a physiological pH range than that of T4. Further structural and mutational studies, as has been performed for the OATP family (28), may help to elucidate the underlying mechanism. Nonetheless, our studies indicate that the mechanism by which SLC17A4 transports T4 in mammalian cells differs from those described for its other putative substrates in proteoliposomes (12).
Of note, TH uptake was lower in DMEM than in saline-based buffers, as has been observed previously for MCT8 and MCT10 (14). We speculate that DMEM may either contain substances that either inhibit SLC17A4, or compete with TH as alternative substrate(s). Alternatively, the abundance of amino acids in DMEM may alter the contribution of endogenous TH transporters (e.g., LATs) to the TH gradient and flux across the cell membrane. As SLC17A4 appears to mediate TH transport through facilitated diffusion, this would directly impact on its activity.
Although we observed a marginal induction of uric acid uptake in SLC17A4-expressing COS-1 cells, uric acid concentrations up to 1000 μM did not inhibit SLC17A4-mediated TH transport. As these levels are well above the reported normal range of uric acid in serum (23), it is not expected that SLC17A4-mediated TH transport is modulated by circulating uric acid concentrations in vivo.
Importantly, the observed 1.4 times induction of uric acid transport in transfected COS-1 cells was less pronounced compared with the reported ∼3 times induction of uric acid uptake in proteoliposomes (12) and may suggest that SLC17A4 is not an efficient uric acid transporter in living cells. These differences may be inherent to the different expression systems, applied buffers, established ion gradients, and differential abundance of redundant transporters in COS-1 cells that may influence the flux of uric acid across the cell membrane.
Our studies also indicated that SLC17A4 is subject to N-linked glycosylation that may contribute to the appearance of different immune-reactive bands around the predicted size of the SLC17A4 monomer. A similar phenomenon was observed on immunoblots for SLC17A3 on the membrane fraction of rat kidney (29). We demonstrated that the inhibition of N-linked glycosylation diminished the 54 kDa band and reduced T4 uptake, suggesting that the ∼54 kDa protein represents the glycosylated active form of SLC17A4.
Our studies suggest that three Asn residues within ECL1 are the most likely targets for N-linked glycosylation, substitution of which by an alanine resulted in a pronounced functional reduction and exclusive appearance of the ∼47 kDa band. Together, these findings support that the ∼47 kDa band represents immature nonglycosylated SLC17A4. In addition, we also observed a prominent band migrating at ∼90–100 kDa, which may correspond with the size of an SLC17A4 dimer. Alternatively, this band may represent a complex with a regulatory protein, given the presence of a PDZ-binding motif in the C-terminal tail of SLC17A4. Further studies are needed to elucidate this feature.
Although the association of the SLC17A4 locus with serum-free T4 concentrations implies that SLC17A4 is involved in TH regulation (10), further studies in Slc17a4 knockout animals or humans carrying pathogenic mutations in SLC17A4 are needed to delineate the physiological role of SLC17A4 in TH homeostasis. Given its predominant expression at mRNA level along the gastrointestinal tract (12), SLC17A4 may be crucial for the absorption of TH from the gut, which comprises a critical step in the enterohepatic cycle of TH (30) and drug delivery in patients treated with levothyroxine (LT4).
Dysfunction of this process could pose a mechanism by which genetic variants in the SLC17A4 locus result in lower serum FT4 concentrations (10). Further studies will detail in which gastrointestinal cells SLC17A4 is expressed and define its subcellular distribution. Since our study showed that SLC17A4 facilitates the cellular uptake and efflux of TH, it may contribute to the transcellular transport of TH at the apical and basolateral membrane.
Further studies should also reveal whether SLC17A4 is able to transport glucuronidated iodothyronines, in particular T4G, which are assumed to comprise important intermediates in the enterohepatic cycle of TH. The ability of SLC17A4 to transport the sulfoconjugated iodothyronines T3S and T4S, which constitute minor metabolites excreted in bile (31), appears limited.
We performed all studies in COS-1 cells, allowing comparison of the basic transporter characteristics of SLC17A4 with those of other transporters previously characterized in our laboratory [e.g., Refs. (6,14,17,18)]. However, it should be noted that the role of SLC17A4 in cellular TH homeostasis under physiological conditions, likely, not only depends on these basic transporter characteristics, but also on its subcellular distribution and the presence of alternative TH transporters [e.g., OATP2B1] (32), both of which may well differ from the situation in transfected COS-1 cells.
Taken together, SLC17A4 facilitates the cellular uptake and efflux of T3 and T4 with great specificity and has a substantially higher affinity for both substrates compared with most other TH transporters identified to date, including MCT8.
Footnotes
Authors' Contributions
S.G., F.S.v.G, Z.C., S.F., and R.E.A.v.H. performed the experiments. S.G., M.E.M., R.P.P., H.H., M.M., and W.E.V. designed the study. S.G., F.S.v.G., and W.E.V. wrote the article. All authors critically reviewed, revised, and approved the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by a grant from the European Thyroid Association (to S.G.) and the Netherlands Organisation for Health Research and Development (project number 113303005) (to W.E.V.).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1