
Abstract
Background:
Maternal thyroid hormone (TH) plays an essential role for fetal development, especially for the cardiovascular system and its central control. However, the precise consequences of altered TH action during the different periods in pregnancy remain poorly understood.
Methods:
To address this question, we used mice heterozygous for a mutant thyroid hormone receptor α1 (TRα1) and wild-type controls that were born to wild-type mothers treated with 3,3′,5-triiodothyronine (T3) during the first or the second half of pregnancy. We then phenotyped the offspring animals as adults by in vivo measurements and postmortem tissue analyses.
Results:
Maternal T3 treatment in either half of the pregnancy did not affect postnatal growth development. Serum thyroxine and hypophyseal thyrotropin subunit beta or deiodinase type II expression was also not affected in any group, only TRα1 mutant males exhibited a reduction in serum T3 levels after the treatment. Likewise, hepatic deiodinase type I was not altered, but serum selenium levels were reduced by the maternal treatment in wild-type offspring of both genders. Most interestingly, a significant increase in heart weight was found in adult wild-types born to mothers that received T3 during the first or second half of pregnancy, while TRα1 mutant males were protected from this effect. Moreover, we detected a significant increase in heart rate selectively in male mice that were exposed to elevated maternal T3 in the second half of the pregnancy.
Conclusion:
Taken together, our findings demonstrate that maternal TH is of particular relevance during the second half of pregnancy for establishing cardiac properties, with specific effects depending on TRα1 or gender. The data advocate routinely monitoring TH levels during pregnancy to avoid adverse cardiac effects in the offspring.
Introduction
Thyroid hormone (TH) is important for development (1). As the fetal thyroid gland does not produce hormones until late pregnancy in humans, around embryonic day 15 in mice (2), the embryonal demand for the hormone needs to be met by the maternal thyroid gland (3). Consequently, defects in maternal thyroid function can severely impact the fetus, especially the developing brain (4), as well as pregnancy outcomes including miscarriage and growth retardation. Moreover, the offspring's setting of the hypothalamic/pituitary/thyroid (HPT) axis can be permanently altered (5,6).
The HPT axis regulates circulating THs via an endocrine feedback loop. Thyrotropin-releasing hormone (TRH) is secreted by the hypothalamus, stimulating the anterior pituitary to produce thyrotropin (TSH), which drives the thyroid gland to release THs. THs in turn negatively regulate TRH and TSH, thereby affecting their own production and release (7). This tightly controlled mechanism is required as TH has potent effects on many physiological functions, including energy homeostasis (8) and cardiac contractility or heart rate (9 –11). The active hormone 3,3′,5-triiodothyronine (T3) acts via nuclear TH receptors (TRs), namely TRα1 and TRβ (12), which bind to target genes and lead to activation or suppression of gene expression (13).
Despite TH's major role in development, it is currently controversial whether pregnant women should be routinely screened for thyroid function, and whether to substitute in subclinical cases (14,15). As maternal thyroid dysfunction can impair cognitive and motor development in the offspring (16 –18), the majority of clinical studies have focused primarily on these endpoints. However, recent clinical and preclinical data have demonstrated that maternal TH is also crucial for the central control of cardiovascular functions in the offspring (19,20), suggesting that these endpoints should also be considered. Nevertheless, the precise consequences of altered gestational TH signaling on the cardiovascular functions of the offspring are still incompletely understood.
Here we used mice heterozygous for the dominant negative TRα1R384C mutation (TRα1+m) in combination with maternal pharmacological T3 treatments to characterize the temporal role of prenatal TRα1 signaling for the fetal programming of cardiovascular properties. Taking advantage of the fact that mutant TRα1 can be reactivated by elevating T3 levels in vivo (21), we set up an experimental paradigm, in which TRα1 signaling in the TRα1+m embryo was selectively restored during the first or second half of pregnancy. The animals were compared with wild-type littermates (TRα1++) exposed to high T3 in the same periods as well as untreated controls.
Our findings show that maternal TH is important for establishing cardiac properties. Especially in the second half of pregnancy, elevated TH levels lead to increased heart rate and weight in the male offspring, associated with permanent changes in pacemaker channel gene expression and DNA methylation.
Materials and Methods
Animal husbandry and study design
Mice were housed at 21°C on a 12-hour light/dark cycle with free access to food and water. Animal procedures were conducted according to EU directives (2010/63/EU) and approved by Ministerium für Energiewende, Landwirtschaft, Umwelt, Natur und Digitalisierung, Schleswig-Holstein, Germany.
Wild-type C57BL/6NCr female mice were purchased at 10 weeks from Charles River, Germany, and mated to heterozygous TRα1+m males to obtain TRα1++ or TRα1+m offspring. Dams were treated either from the first day after positive plug (E0.5) until E12.5 or from E12.5 until E18.5 with T3 via drinking water (0.5 μg/mL; T6397, Sigma Aldrich, Germany) containing 0.01% BSA (A7906, Sigma Aldrich). If not stated otherwise, adult offspring were analyzed at four to five months.
Gene expression measurement by quantitative real-time PCR
RNA was isolated using the RNeasy mini or fibrous tissue mini kit (QIAGEN, Germany) including a DNase digestion step to prevent genomic DNA contamination. cDNA was synthesized of 1 μg RNA using the Molecular Biology RevertAid cDNA Kit (Thermo Fisher Scientific, Germany). QuantStudio (Thermo Fisher Scientific, Germany) and GoTaq® qPCR Master Mix (Promega, Germany) were used for quantitative real-time PCR (qPCR). Reference genes were determined using NormFinder (
Heart rate and blood pressure analysis
For blood pressure measurements, a noninvasive tail-cuff system with a temperature-controlled platform (100°F) was used (Model SC-100; Hatteras Instrument Incorporated, USA). Blood pressure (diastolic, systolic, and mean arterial pressure) was measured 15 times in 1 session and averaged over 3 consecutive days.
Serum parameter measurements: total T3 and thyroxine enzyme linked immunosorbent assay
Serum concentrations of total T3 and total thyroxine (T4) were determined by enzyme linked immunosorbent assay (ELISA) (tT3: DNOVO53, NovaTec Immundiagnostica GmbH, Dietzenbach, Germany, detection limit 0.05 ng/mL, intra- and interassay variance <11%; tT4: ELISA, EIA-1781; DRG Instruments GmbH, Marburg, Germany, detection limit 0.5 ng/mL, intra- and interassay variance <4.5%).
Western blot analysis
Hearts were homogenized in RIPA buffer with protease inhibitors (Roche Diagnostics GmbH, Germany). Fifty micrograms of protein were separated on a 12% sodium duodecyl sulfate-polyacrylamide gel (Bio-Rad Laboratories, Germany), transferred onto a membrane (PVDF; Merck Millipore, Germany), and probed with the rabbit polyclonal anti-HCN2 (1:1000 dilution; APC-30, Alomone, Israel) or rabbit polyclonal anti-HCN4 antibodies (1:1000 dilution; APC-052, Alomone, Israel) followed by a peroxidase-conjugated secondary goat anti-rabbit-IgG antibody (P0448, DAKO, Denmark; 1:5000 dilution), visualized using an ECL system (Chemi Doc Touch; Bio-Rad Laboratories, Germany), and normalized to total protein.
DIO1 and GPX1 activity assays
DIO1 activity was analyzed by a nonradioactive assay (22). Briefly, liver homogenates were mixed with dimethylsulfoxide or propylthiouracil (PTU), and incubated with a substrate mix at 37°C. The released iodide was determined by a Sandell/Kolthoff reaction and the background from PTU-inhibited controls subtracted. Interassay variance was below 20%. GPX1 activity was determined by a coupled enzymatic assay (23). Inter- and intra-assay coefficient of variability was below 15%.
Trace element measurements
Total concentrations of copper and selenium in serum and liver homogenates were determined by total reflection X-ray fluorescence using a benchtop analyzer (S4 T-STAR; Bruker Nano GmbH, Berlin, Germany), as described (24), with a Seronorm standard (Sero AS, Billingstad, Norway).
DNA methylation by bisulfite pyrosequencing
Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN), quantified (Quantus Fluorometer, Promega, Germany), and converted with sodium-bisulfite using the EpiTect Fast DNA Bisulfite Kit (QIAGEN). PCR was performed using the PyroMark PCR Kit (QIAGEN) and primers generated with the PyroMark Assay Design Software 2.0 (QIAGEN). The PCR product was pyrosequenced on a PyroMark Q48 Autoprep with Q48 Advanced CpG Reagents (QIAGEN). A standard curve of 0%, 25%, 50%, 75%, and 100% methylated DNA was used to quantify absolute CpG methylation.
Statistical analysis
Data were analyzed using two-way analysis of variance (ANOVA) with offspring genotype and maternal treatment as independent variables followed by Holm/Sidak's multiple comparison test using Prism 7 software (GraphPad Software, Inc., San Diego, CA, USA). Males and females were tested separately. Values are mean ± standard error of the mean. Statistical significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, and details can be found in Supplementary Table S1.
Result
Experimental setup
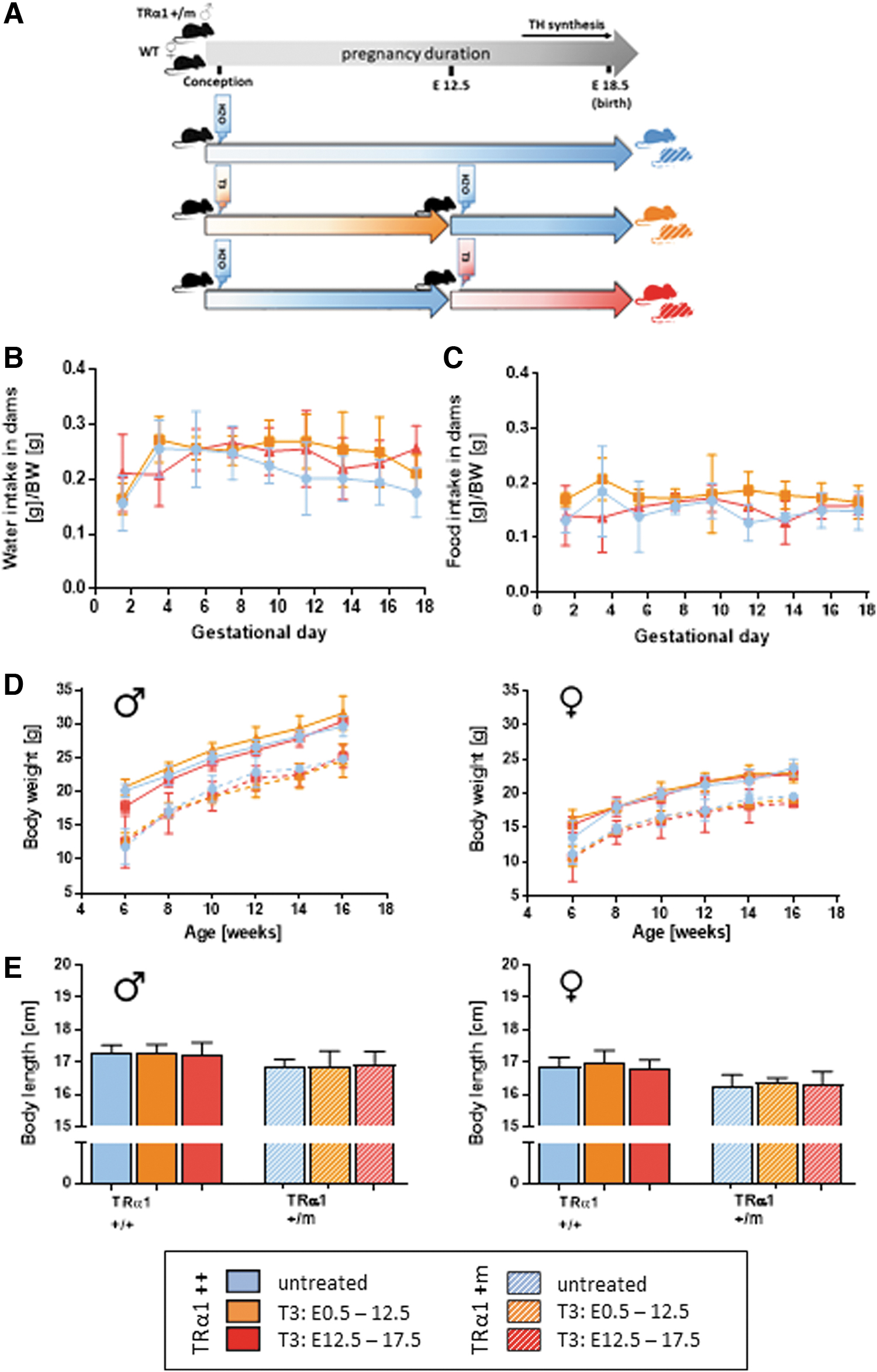
We used mice TRα1+m mice born to wild-type mothers as model system for impaired TH signaling in utero. As this particular mutation can be reactivated by elevating TH levels (21,25), we treated the mother with 0.5 mg/L T3 in drinking water either in the first or in the second half of pregnancy (Fig. 1A), a treatment shown to result in 8–10-fold elevated serum T3 levels in adult animals and sufficient to restore embryonal TRα1 signaling selectively in these periods (4,26 –28). As the respective TRα1++ littermates were exposed to elevated TH during the same periods, we obtained the entire range of impaired or enhanced TH signaling in the developing embryos.
(
T3 treatment has no effect on offspring's development
Throughout pregnancy, daily water intake per body weight was comparable with nonpregnant adult mice, indicating sufficiently high daily oral T3 uptake to reactivate the mutant TRα1 in utero (4,26 –28), with no effect of the T3 treatment on dams' water and food intake (Fig. 1B, C). Litter sizes were comparable (untreated [n = 5]: 7.4 ± 2.4; T3 E0.5–12.5 [n = 6]: 8.0 ± 1.7; T3 E12.5–18.5 [n = 7]: 9.0 ± 1.2). Postnatal weight development was not affected by maternal T3 treatment (Fig. 1D; Supplementary Table S1); however, body weight was lower in TRα1+m as expected (21). Total body length at the age of 16 weeks reflected this pattern (Fig. 1E).
T3 treatment during pregnancy decreases offspring's serum selenium
Maternal thyroid disease can affect the HPT-axis setting (29), however, our maternal T3 treatment caused reduced T3 only in the adult TRα1+m male offspring (Fig. 2A; Supplementary Table S1). Neither serum total T4 (Fig. 2B) nor pituitary thyrotropin subunit beta (Tshb) or deiodinase type II (Dio2) mRNA expression was altered (Fig. 2C, D), suggesting that the HPT-axis was largely unaffected. Deiodinase type I (Dio1) mRNA and DIO1 activity were higher in TRα1+m as expected (30); however, changes by maternal treatment in either period were not significant (Fig. 2E, F). Maternal T3 reduced Spot14 mRNA in female but not male offspring (Fig. 2G), while hepatic GPX1 activity, as well as selenium and copper levels, were not altered in either condition (Supplementary Fig. S1A; Fig. 2H), concurring with previous studies (31). Remarkably, serum selenium levels were strongly reduced in males of both genotypes and wild-type females upon either maternal T3 treatment (Fig. 2I), while serum copper levels, also regulated by TH (32), were not altered (Fig. 2J).

(
Gender- and genotype-specific cardiovascular alterations in the offspring
As maternal TH can affect the cardiovascular set point (4,20), we analyzed adult offspring using a noninvasive tail-cuff system. T3 treatment in the second half of pregnancy led to a significant increase of heart rate in wild-type and TRα1+m male offspring, while not affecting wild-type females and even showing the reverse in TRα1+m females (Fig. 3A; Supplementary Table S1). Heart rate was generally lower in TRα1+m consistent with previous studies (33 –35). Blood pressure was unaffected except for a mild increase in wild-type males and a decrease in TRα1+m females treated in the second half of pregnancy (Fig. 3B–D). Remarkably, wild-type male and female offspring displayed significant hypertrophy when mothers were treated in the second half of pregnancy (Fig. 3E). While heart weight was lower in TRα1+m as expected, it was further lowered by maternal T3 treatment selectively in males.

(
Cardiac gene expression after maternal T3 treatment
Maternal T3 treatment led to a significant upregulation of cardiac alpha myosin heavy chain (α-Mhc), cardiac beta myosin heavy chain (β-Mhc) (35 –37), hyperpolarization-activated cyclic nucleotide-gated channel 2 and 4 (Hcn2 and Hcn4), and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (Serca2) in adult male offspring of both genotypes (Fig. 4A–G; Supplementary Table S1), with the exception of Hcn2, which was only affected by T3 in the second half of pregnancy. Females were less affected, primarily TRa1+m offspring treated in the second half of pregnancy—an effect not caused by protective deiodinase III (Dio3) expression (Fig. 4H).

(
Maternal T3 treatment in the second half of pregnancy leads to altered DNA methylation in Hcn2 and Hcn4 genes
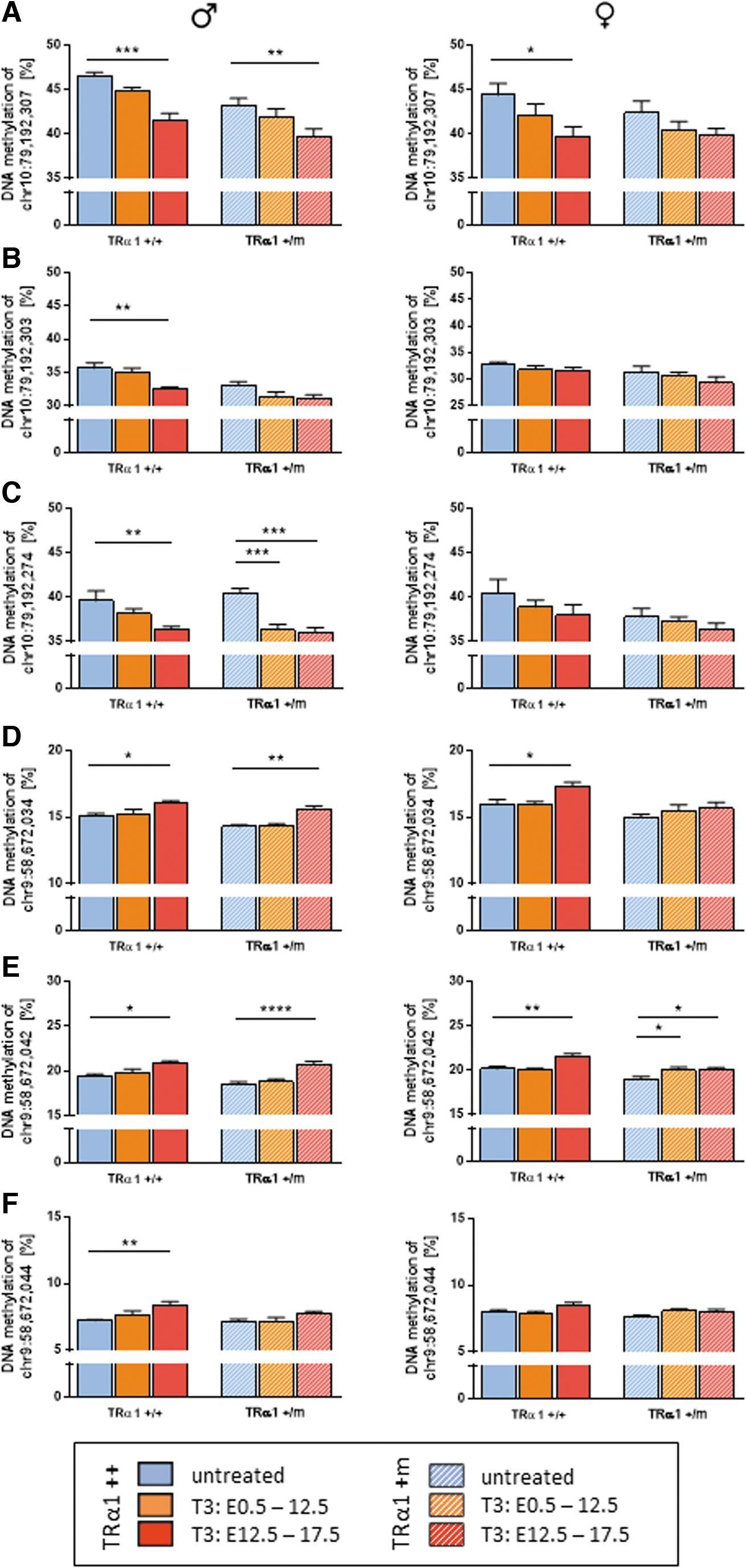
Given that pacemaker Hcn2 and Hcn4 expression correlated strongly with the heart rate (Supplementary Fig. S1C, D, and Supplementary Table S1), we hypothesized that altered fetal programming by DNA methylation in these genes could underlie the persistently elevated expression. Using the publicly available genome-wide methylation data sets, GSE21415 (38) and ENCSR397YEG (39), we identified CpG sites in these genes that are actively methylated/demethylated during cardiac development, expecting that they might be most sensitive to reprogramming by maternal TH. Subsequently, we measured DNA methylation in intron four of the Hcn2 gene, observing a consistent decrease in three of five CpG sites in male offspring of both genotypes induced by T3 treatment in the second half of pregnancy (Fig. 5A–C; Supplementary Fig. S1E, F, and Supplementary Table S1). Within exon 1 of Hcn4, three of the four CpG sites showed a significant increase in DNA methylation after T3 treatment in the second half of pregnancy (Fig. 5D–F; Supplementary Fig. S1G). For the affected CpG sites, Hcn2 expression correlated negatively and Hcn4 positively with DNA methylation (Supplementary Fig. S2), suggesting an altered chromatin state in these regulatory regions.
(
Discussion
Several studies have shown that maternal TH affects embryonal development (40,41) and particularly the cardiovascular system (20,42,43). However, the precise window of TH action and its mechanistic targets have remained unclear. Our study now provides a phenotypical roadmap on the cardiovascular consequences of altered TH signaling during the first or second half of murine pregnancy, corresponding to the first and second trimester in humans.
Consequences of maternal T3 treatment on early development and HPT axis
While altered TH levels during the entire pregnancy can affect body mass and body length of the offspring (44,45), this was not observed in our study, likely due to the shorter treatment being insufficient to induce catabolic conditions. TRα1+m offspring were lighter than their control littermates as expected (21), underlining the well-established importance of TRα1 for postnatal development (46,47).
Although maternal TH can interfere with the offspring's HPT axis, this was generally not observed in our paradigm. Likewise, Dio1 as a sensitive hepatic T3 marker was not affected by maternal T3 treatment, and only elevated in TRα1+m mice as observed previously (30). However, there was an interesting tendency of increased Dio1 mRNA and activity despite the somewhat reduced serum TH levels in the maternally T3-treated offspring. This may suggest altered hepatic programming, concurring with a study revealing that altered perinatal hepatic TH action by abolishing Dio2 expression leads to permanent epigenetic modifications in the liver (48).
Interestingly, serum selenium levels were reduced in wild-type offspring upon maternal T3 treatment in the second half of pregnancy. While T3 is known to positively regulate serum selenium levels in the adult animal (31), our findings rather suggest that selenium metabolism may also be epigenetically reprogrammed by maternal T3. A similar effect was observed in congenital hypothyroid children (49). Despite sufficient T4 substitution, their serum selenium levels remained lower than controls, suggesting that even a minor transient developmental hypothyroidism can cause permanent alterations in adult selenoprotein physiology. As hepatic selenium levels, DIO1, and GPX1 enzyme activity were unaffected, the liver is likely not the origin of the effect; therefore, further studies addressing this are warranted.
Sexual dimorphic effects on cardiac functions after maternal T3
Although variations in maternal TH function affect cardiovascular parameters in humans (42) and mice (4,20), blood pressure was generally unaffected. In contrast, heart weight was increased in wild-types of mothers treated in the second half of pregnancy with T3; an effect that was abolished in male TRα1+m offspring, suggesting a protective role of the mutant TRα1. This concurs with previous studies showing reduced heart weight in the offspring of hypothyroid mothers (50), and the stronger phenotype in offspring exposed to high T3 in the second half of pregnancy matches with the onset of TRα1 expression at E13.5 in mice (51). While the precise reason for the hypertrophy remains to be established, it can be speculated that maternal T3 either acts in the developing fetal brain, given that a central mechanism also drives cardiac hypertrophy in adult hyperthyroidism (52), or directly in the fetal heart. However, we found elevated α- and β-Mhc in the treated offspring, although these genes are usually oppositely regulated by T3. Together with the fact that hyperthyroidism-induced hypertrophy is usually reversed after reestablishing euthyroidism (53), the precise mechanism seems to be different than the classic cardiac T3 signaling cascades.
In contrast to the hypertrophy, which was similar in males and females, we observed a sexual dimorphism in heart rate: Male offspring of mothers treated with T3 in the second half of pregnancy had increased heart rate in both genotypes, although heart rate was generally lower in TRα1+m mice as expected (33), while a tachycardia was absent in females. This effect correlated with the level of pacemaker gene expression, especially HCN4 protein, suggesting that these genes may drive the tachycardia. Why the effect was only apparent in males remains unknown, but it could be speculated that the previously described cross talk between TH and sex hormones (54) or the well-known sexual dimorphism in deiodinase regulation (55) might contribute to differences in local TH action during cardiac development.
Epigenetic alterations in pacemaker genes associated with maternal T3
Based on the persistently elevated level of Hcn2 and Hcn4 gene expression, we hypothesized that altered fetal programming by DNA methylation might contribute to the observed phenotype. This concept of an interaction between TH and epigenetic programming has been established recently in several elegant studies, which demonstrated that (i) Dio3 is an important gatekeeper for epigenetic programming in the brain (56), (ii) the sensitivity to TH action in the offspring is also epigenetically controlled (57), and (iii) alterations in maternal TH indeed affect DNA methylation patterns of several genes (58). It is therefore not surprising that we observed a similar effect of maternal TH on DNA methylation of Hcn2 and Hcn4, especially since intronic regions are known to be of regulatory importance for the regulation of Hcn genes (59). While some CpG sites were affected in both genders, others were affected only in males, an effect often observed in fetal programming (60). T3 treatment in the second half of pregnancy had a more pronounced effect on DNA methylation, concurring with the important role of TRα1 in this developmental period of the heart (61). It remains to be elucidated whether these DNA methylation changes indeed causally affect gene expression, and how far increased HCN2 and HCN4 contribute to tachycardia in males.
Conclusion
In conclusion, our study reveals that maternal TH signaling—in particular in the second half of pregnancy—is important for establishing cardiac properties in the offspring. These findings are of clinical relevance, as a large proportion of pregnant women display thyroid disorders (62), but there is a controversial discussion about whether pregnant women should be routinely tested (63). Unfortunately, the current arguments are largely based on studies with psychological and motoric endpoints. Our study now advocates for the inclusion of cardiovascular endpoints in these studies and underlines the necessity to study endocrine disrupting compounds during pregnancy (64).
Footnotes
Acknowledgments
We thank the staff of the animal facility (GTH) of the University of Lübeck for their help. The support of Cathleen Geissler and Henriette Kirchner with the epigenetic quantifications is greatly appreciated.
Authors' Contributions
M.P., R.O., Q.S., and J.R. performed the experiments; L.S. and J.M. designed the research and supervised the experiments; M.P. and J.M. drafted the article; and all authors edited and approved the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the German Research Foundation DFG (grant numbers MI1242/3-2 to J.M. and OE723/2-1 to R.O.), the GRK1957 “Adipocyte-Brain-Crosstalk,” the TR-SFB296 “LocoTact” (P12, P14, and P17 to J.M. and L.S.), and Research Unit FOR-2558 “TraceAge” (Scho 849/6-2 to L.S.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1