
Abstract
Background:
The sodium/iodide symporter (NIS) mediates active iodide accumulation in the thyroid follicular cell. Autosomal recessive iodide transport defect (ITD)-causing loss-of-function NIS variants lead to dyshormonogenic congenital hypothyroidism due to deficient iodide accumulation for thyroid hormonogenesis. Here, we aimed to identify, and if so to functionally characterize, novel ITD-causing NIS pathogenic variants in a patient diagnosed with severe dyshormonogenic congenital hypothyroidism due to a defect in iodide accumulation in the thyroid follicular cell, as suggested by nondetectable radioiodide accumulation in a normally located thyroid gland, as well as in salivary glands.
Methods:
The proposita NIS-coding SLC5A5 gene was sequenced using Sanger sequencing. In silico analysis and functional in vitro characterization of the novel NIS variants were performed.
Results:
Sanger sequencing revealed novel compound heterozygous SLC5A5 gene variants (c.970-3C>A and c.1106A>T, p.D369V). In silico analysis suggested that c.970-3C>A disrupts the canonical splice acceptor site located in intron 7. Splicing minigene reporter assay revealed that c.970-3C>A causes exon 8 skipping during NIS pre-mRNA splicing leading to the NIS pathogenic variant p.Y324Hfs*148. Moreover, in silico analysis indicated p.D369V as pathogenic. Functional in vitro studies demonstrated that p.D369V NIS does not mediate iodide accumulation, as p.D369V causes NIS to be retained in the endoplasmic reticulum. Mechanistically, we propose an intramolecular ionic interaction involving the β carboxyl group of D369 and the guanidinium group of R130, located in transmembrane segment 4. Of note, an Asp residue at position 369—which is highly conserved in SLC5A family members—is required for functional NIS expression at the plasma membrane.
Conclusions:
We uncovered a critical intramolecular interaction between R130 and D369 required for NIS maturation and plasma membrane expression. Moreover, we identified the first intronic variant causing aberrant NIS pre-mRNA splicing, thus expanding the mutational landscape in the SLC5A5 gene leading to dyshormonogenic congenital hypothyroidism.
Introduction
Iodide transport defect (ITD) (Online Mendelian Inheritance in Man #274400) is an uncommon autosomal recessive disorder where the thyroid follicular cell cannot accumulate the iodide required for thyroid hormonogenesis, thus leading to dyshormonogenic congenital hypothyroidism (1). Diagnostic hallmarks of this disease are reduced to absent radioiodide accumulation in a normally located thyroid gland and a low saliva-to-plasma iodide ratio.
Iodide transport is the first step in the biosynthesis of the iodine-containing thyroid hormones. The sodium/iodide symporter (NIS) is a plasma membrane glycoprotein that mediates active iodide accumulation in the thyroid as well as other tissues, such as salivary glands and lactating breast (2). In the thyroid, NIS expression is restricted to the basolateral surface mediating iodide transport from the bloodstream into the thyroid follicular cells. We uncovered a conserved monoleucine-based sorting motif located in the carboxy-terminus of NIS that constitutes a sorting signal required for basolateral expression (3). Moreover, recently, we identified a carboxy-terminal PDZ-binding motif that participates in the polarized expression of NIS by selectively interacting with the PDZ-domain containing protein SCRIB, thus retaining the transporter at the basolateral plasma membrane (4).
To date, over 30 pathogenic variants in the NIS-coding SLC5A5 gene have been identified in patients with thyroid dyshormonogenesis. The detailed molecular analysis of several ITD-causing NIS variants has revealed critical amino acids for substrate binding, specificity, and stoichiometry, as well as for folding and plasma membrane targeting (5). Recently, we reported a molecular characterization of the p.G561E NIS variant, which impairs the recognition of an adjacent tryptophan-acidic motif by the kinesin-1 subunit kinesin light chain 2, interfering with NIS maturation beyond the endoplasmic reticulum, and reducing iodide accumulation (6). Moreover, based on structure-function analysis of pathogenic NIS variants, we developed a machine learning-based NIS-specific variant classifier to improve the prediction of pathogenicity of missense NIS variants in clinical practice (7).
Here, we report a detailed functional characterization of novel compound heterozygous SLC5A5 pathogenic variants (c.970-3C>A and c.1106A>T, p.D369V), identified in a pediatric patient with severe dyshormonogenic congenital hypothyroidism caused by deficient iodide accumulation. We show that c.970-3C>A disrupts the canonical splice acceptor site located in intron 7 leading to exon 8 skipping during NIS pre-mRNA splicing, whereas the missense variant p.D369V abrogates iodide accumulation due to retention of NIS in the endoplasmic reticulum. Mechanistically, we propose that p.D369V impairs an intramolecular ionic interaction involving the β carboxyl group of D369 and the guanidinium group of R130, located in transmembrane segment (TMS) 4, which is critical for the proper folding required for NIS exit from the endoplasmic reticulum.
Materials and Methods
Patient
The proposita was a post-term (42 weeks) female infant born as the only child of biochemically euthyroid, nonconsanguineous Caucasian (of European descent) parents. On day 11, an abnormally high thyrotropin (TSH) level was detected by newborn screening (>200 μU/mL, cutoff 15 μU/mL). The diagnosis of congenital hypothyroidism was confirmed by serum TSH >40 μU/mL (1.3–10 μU/mL), total thyroxine (T4) 1 μg/dL (6.0–14.0 μg/dL), and total triiodothyronine (T3) 30 ng/dL (80–240 ng/dL). Thyroid autoantibodies were negative. Thyroglobulin measurement was not available.
Neonatal clinical evaluation revealed tongue protrusion, jaundice, and hoarse cry. Knee X-ray revealed reduced ossification of the distal femoral epiphyseal nucleus. Thyroid hormone supplementation was started at day 17 (50 μg/day levothyroxine) with excellent treatment adherence during the first years of life. Levothyroxine dosage was adjusted regularly based on thyroid function tests. At 2.5 years of age, after levothyroxine withdrawal for a month, thyroid function evaluation indicated permanent disease (TSH >65 μU/mL, total T4 1 μg/dL, and total T3 30 ng/mL). Ultrasonography showed a normal size and properly located thyroid gland. Radionuclide scintigraphy revealed nondetectable 131I-iodide accumulation in the thyroid gland, as well as salivary glands. At 8 years of age, evaluation using the WISC-III test demonstrated a total intelligence quotient of 84, with slow performance in visuospatial skills.
Thyroid function tests
Thyroid function analyses were performed by DELFIA system (PerkinElmer, Waltham, MA).
Sanger sequencing
The genetic analysis was approved by the Ethics Committee of the Hospital de Niños Dr. Ricardo Gutierrez and performed under informed written consent of the proposita's mother. Genomic DNA was extracted from peripheral blood mononuclear cells using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI). The nucleotide sequence of all coding exons and exon/intron boundaries of the SLC5A5 gene was determined by Sanger sequencing by capillary electrophoresis (Macrogen, Seoul, South Korea) (8).
Expression vectors and site-directed mutagenesis
The cloning of the amino-terminus hemagglutinin (HA)-tagged human NIS expression vector was described previously (9). Site-directed mutagenesis was performed with oligonucleotides carrying the desired mutation using Phusion Hot Start II DNA Polymerase (Thermo Fisher Scientific, Waltham, MA) (10). All constructs were sequenced to verify specific nucleotide substitutions (Macrogen).
Cell culture and transfections
Human embryonic kidney (HEK)-293T cells (CRL-3216) and Fisher rat-derived thyroid cells FRTL-5 (CRL-1468) were obtained from the American Type Culture Collection (Rockville, MD) and cultured as described previously (11,12). Cells were transiently transfected at a ratio of 4 μg plasmid/10 cm dish using Lipofectamine 2000 (Thermo Fisher Scientific) (13).
Splicing minigene reporter assays
Minigene constructs containing a fragment of the SLC5A5 gene encompassing 160 nucleotides of wild-type (WT) or c.970-3C>A intron 7, the 89 nucleotides encoding the exon 8, and 160 nucleotides of intron 8 were synthesized (Macrogen) and subcloned into the EcoRI and BamHI cloning sites of the splicing reporter pSPL3 vector (a discontinued product of Thermo Fisher Scientific) (14). Total RNA was extracted 24 hours after transfection with pSPL3-based minigene reporters using the Direct-zol RNA MiniPrep Kit (Zymo Research, Irvine, CA) (15). Complementary DNA synthesis and polymerase chain reaction (PCR) were performed as described (16). The pSPL3-specific primer sets were as follows: SD2 5′-GTGAACTGCACTGTGACAAGCTGC and SA4 5′-CACCTGAGGAGTGAATTGGTCG. Reverse transcription polymerase chain reaction products were resolved by electrophoresis on 2.5% agarose gels containing ethidium bromide.
125I-iodide transport assays
Cells seeded onto poly-
Flow cytometry
Cells were fixed in 2% paraformaldehyde and stained with 0.4 μg/mL mouse monoclonal anti-HA-Tag (sc-7392; Santa Cruz Biotechnology, Santa Cruz, CA) antibody in phosphate-buffered saline (PBS) containing 0.2% human serum albumin for nonpermeabilized conditions, or an additional 0.2% saponin for permeabilized conditions (18). After washing, cells were incubated with 1 μg/mL Alexa-488-conjugated anti-mouse antibody (A-11029; Molecular Probes, Eugene, OR). The fluorescence of ∼5 × 104 events per tube was assayed in a BD FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA). Data analysis was performed with FlowJo software (Tree Star, Ashland, OR). Percent mean fluorescent intensity (%MFI) was calculated as (MFI mutant NIS)*100/(MFI WT NIS).
Immunofluorescence
Cells seeded onto poly-
Western blot
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), electrotransference to nitrocellulose membranes, and immunoblotting were performed as previously described (21). Membranes were blocked and incubated with 0.2 μg/mL affinity-purified rabbit polyclonal anti-human NIS and 0.1 μg/mL mouse monoclonal anti-α-tubulin (T9026; Sigma-Aldrich, St. Louis, MO) primary antibodies. After washing, membranes were incubated with 0.07 μg/mL IRDye 680RD goat anti-rabbit (#926-68071) and IRDye 800CW goat anti-mouse (#926-32210) secondary antibodies (LI-COR Biosciences, Lincoln, NE). Membranes were visualized by the Odyssey Infrared Imaging System (LI-COR Biosciences).
Statistical analysis
Results are presented as the mean ± standard error of the mean of three independent experiments. Statistical tests were performed using Prism 5.0 software (GraphPad Software, La Jolla, CA). Multiple group analysis was conducted by one-way ANOVA and Newman–Keuls multiple-comparisons post hoc test. Comparisons between two groups were made using paired Student's t-test. Differences were considered significant at p < 0.05.
Results
Identification of novel compound heterozygous SLC5A5 variants
ITD was suspected in the proposita based on dyshormonogenic congenital hypothyroidism and nondetectable radioiodide accumulation in a normally located thyroid gland, as well as in salivary glands. Proposita's SLC5A5 gene sequencing revealed novel compound heterozygous variants: c.970-3C>A (intron 7) and c.1106A>T (exon 9), p.D369V (RefSeq: NM_000453) (Fig. 1A). The variant c.970-3C>A has been reported in the Single Nucleotide Polymorphism Database (rs781024695) in heterozygosis showing a minor allele frequency of 0.000003984, according to The Genome Aggregation Database. The variant p.D369V was not reported in public databases. Consistent with the recessive nature of the disease, the euthyroid mother was a heterozygous carrier of the c.970-3C>A variant (Fig. 1B). The father was unavailable for genetic study.
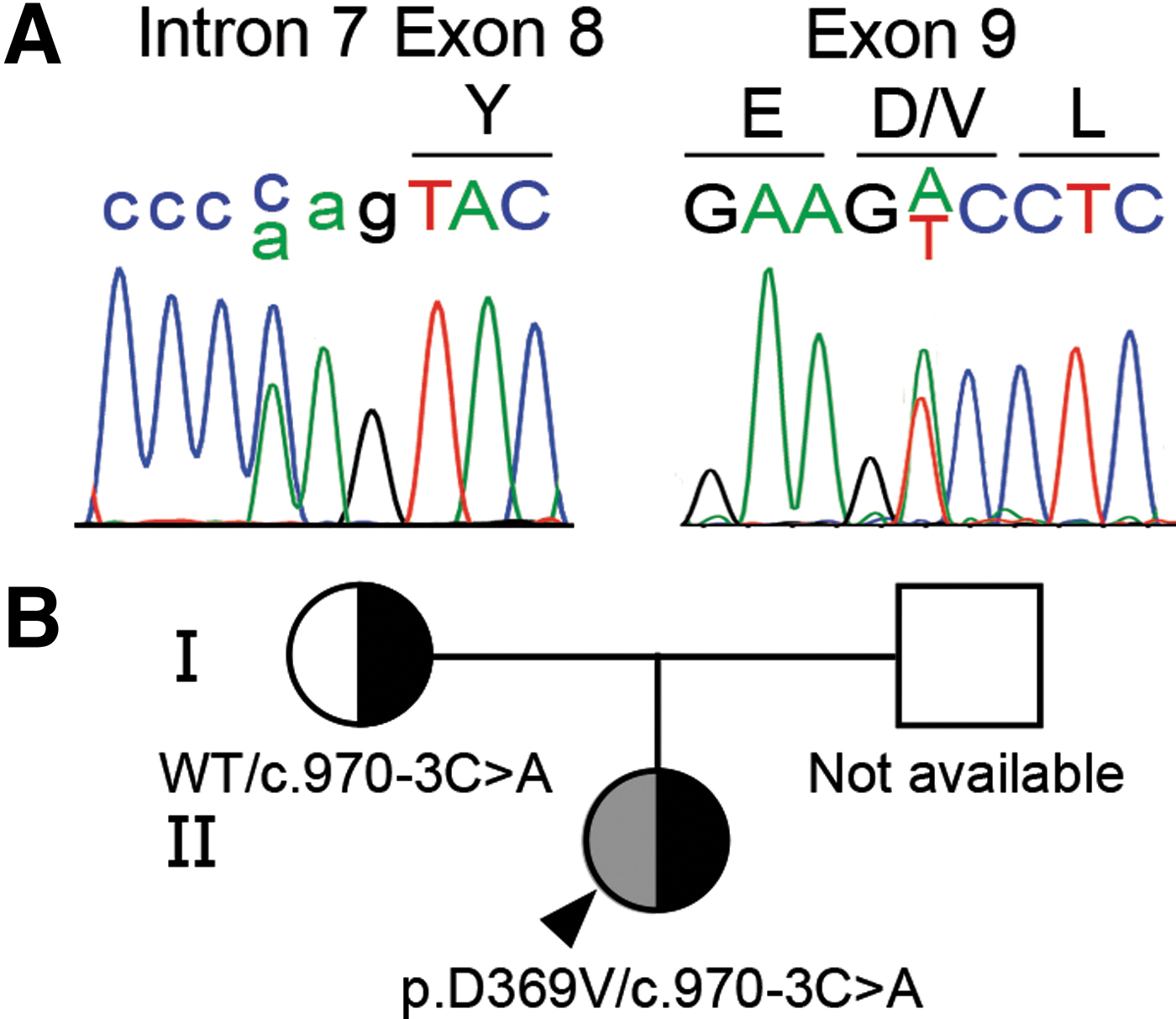
Identification of novel SLC5A5 pathogenic variants causing dyshormonogenic congenital hypothyroidism. (
The pathogenic variant c.970-3C>A impairs normal NIS pre-messenger RNA splicing
In silico analysis using splice site prediction software revealed that c.970-3C>A disrupts the canonical splice acceptor site located at the boundary of intron 7 and exon 8, thus affecting normal exon 8 inclusion during NIS premessenger RNA splicing (Table 1). To functionally assess the impact of the variant c.970-3C>A, pSPL3-based minigene constructs were generated and tested in transiently transfected FRTL-5 thyroid cells (Fig. 2A). Consistently with in silico prediction software, the minigene assay revealed that the variant c.970-3C>A generates one major transcript of 153 bp, compatible in size with the skipping of exon 8 (β splicing product), as a similar pattern was observed when cells were transfected with the empty reporter vector (Fig. 2B).

The variant c.970-3C>A causes mis-splicing of exon 8. (
In Silico Analysis of c.970-3C>A Sodium/Iodide Symporter Using Splicing Prediction Algorithms
The variant 970-3C>A, located at position -3 in the splice acceptor site of intron 7, is underlined. Intron 7 is shown in lowercase letters and exon 8 in uppercase. Scores obtained for WT and 970-3C>A intron 7 splice acceptor sites using the indicated software. For each software, scale range is indicated in brackets. The deleterious effect of the variant is predicted if the change in variation was greater than 15% (% variation = [(Variant score − WT score)/WT score]*100).
ASSP, alternative splice site predictor; HSF, human splicing finder; MES, max entropy scan; ND, splice acceptor site was not detected; NNSplice, splice site prediction by neural network; SSF-like, splice site finder-like; WT, wild type.
By contrast, the WT minigene generates one major transcript of 242 bp compatible with the canonical spliced transcript (α splicing product) (Fig. 2B). Significantly, c.970-3C>A variant-caused exon 8 skipping changes the open reading frame of the transcript and generates a downstream premature translation stop codon, leading to the NIS pathogenic variant p.Y324Hfs*148. However, the algorithm NMDEscPredictor (22) predicted that the transcript encoding the frameshift variant p.Y324Hfs*148 undergoes nonsense-mediated decay.
The variant p.D369V NIS is intracellularly retained
According to the rat NIS homology model based on the crystal structure of the Vibrio parahaemolyticus sodium/galactose symporter (vSGLT) (23), a homologue of the eukaryotic sodium/glucose transporter 1 (SGLT1), the residue D369 is located close to the cytoplasmic end of the TMS9 (Fig. 3A). In silico analysis predicted the variant p.D369V as pathogenic (Table 2). Functional analysis revealed that HEK-293T cells, which do not express NIS endogenously, transiently transfected to express that p.D369V NIS displayed undetectable perchlorate-sensitive iodide uptake compared with WT NIS-expressing cells (Fig. 3B). Flow cytometry analysis of plasma membrane NIS expression showed that the levels of p.D369V NIS were significantly lower than those of WT NIS, suggesting that plasma membrane sorting of the mutant protein is severely impaired (Fig. 3C, surface).

The variant p.D369V NIS is retained in the endoplasmic reticulum. (
In Silico Analysis of p.D369V Sodium/Iodide Symporter Using Prediction Algorithms
In silico predictions were carried out using PolyPhen-2 (scale: 0 = benign, 1 = probably damaging), MutationTaster2 (p-value for prediction confidence: 0 = low confidence, 1 = high confidence), and SIFT (scale: 1 = tolerated, 0 = deleterious).
PolyPhen-2, polymorphism phenotyping v2; SIFT, sorting intolerant from tolerant.
By contrast, the analysis of total NIS expression showed similar levels for both proteins (Fig. 3C, total). On Western blots, the electrophoretic pattern of p.D369V NIS showed nonglycosylated (∼55 kDa, band A) and immaturely glycosylated (∼60 kDa, band B) polypeptides, indicating that the mutant protein exhibits only partial maturation (Fig. 3D). By contrast, the plasma membrane-located fully glycosylated (∼100 kDa, band C) NIS polypeptide was only detected in cells expressing WT NIS (Fig. 3D). Immunofluorescence confocal microscopy colocalization experiments with the endogenous endoplasmic reticulum marker retention signal KDEL revealed that p.D369V NIS was retained in the endoplasmic reticulum (Fig. 3E, top panel).
Complementarily, we validated our observations in FRTL-5 thyroid cells transiently transfected to express WT or p.D369V NIS. Immunofluorescence analysis showed that p.D369V NIS is mostly retained in the endoplasmic reticulum (Fig. 3E, lower panel). Interestingly, multiple sequence alignment of NIS orthologues from different metazoan species revealed that the missense variant p.D369V affects a fully conserved residue (Fig. 3F), supporting its importance in NIS processing. Moreover, aspartic acid residues at the position corresponding to D369 in the NIS are highly conserved in SLC5A family members (Fig. 3G).
The interaction between R130 and D369 is required for proper NIS transport to the plasma membrane
To understand the importance of the negative charged residue Asp at position 369, we searched for putative interaction partners in the current NIS homology model (23). In the inwardly open conformation, D369 lies in a highly polar region of NIS formed by TMS4 and TMS9 that faces the cytoplasm. Thus, a hydrophobic residue at this position—such as Val in p.D369V NIS—probably alters the interaction between TMS4 and TMS9, preventing the protein from being properly folded. Based on the model, we predicted an ionic interaction involving the β carboxyl group of D369 and the guanidinium group of R130 located in TMS4 (Fig. 4A).
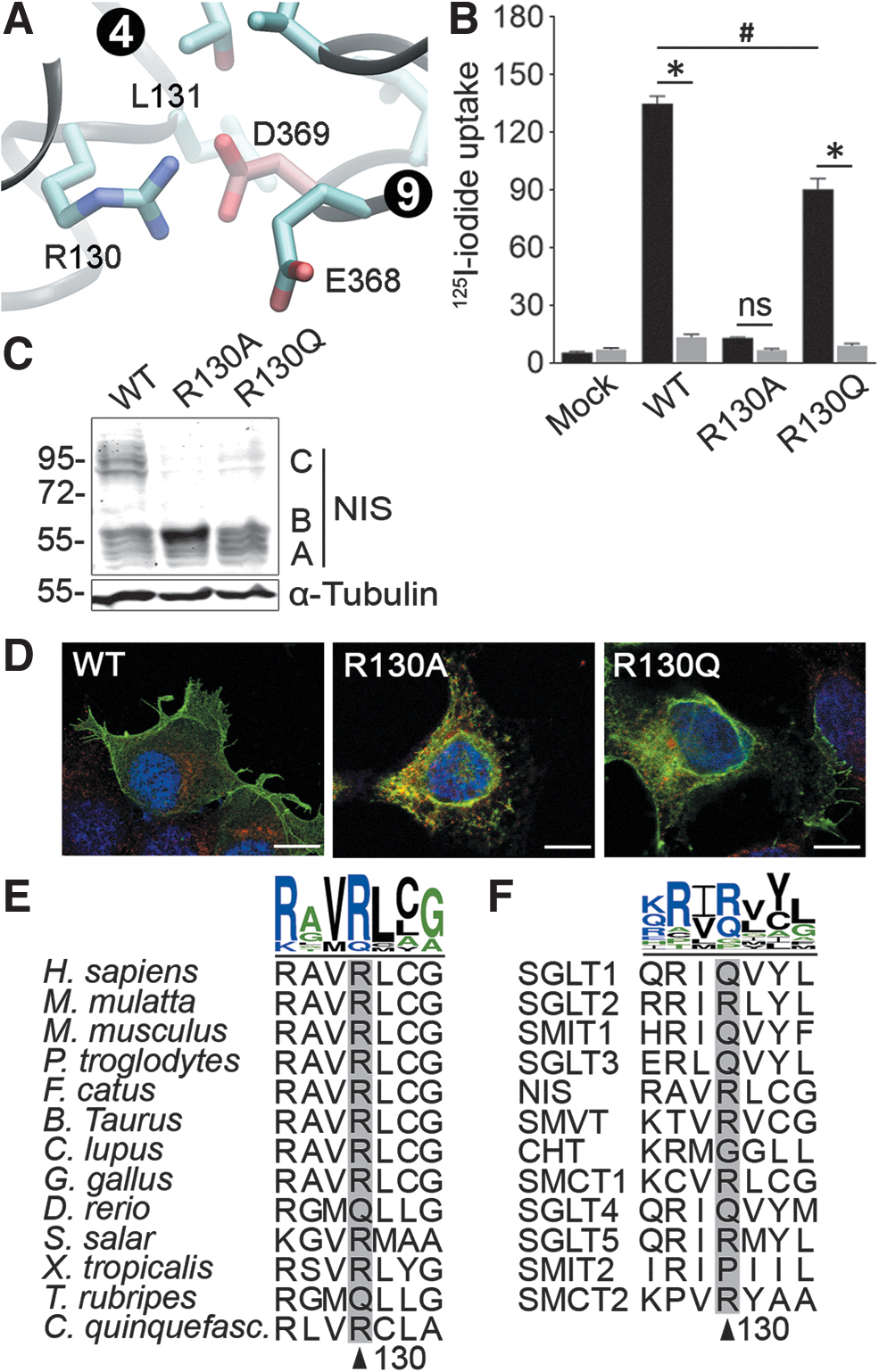
R130 is required for NIS transport to the plasma membrane. (
To test this hypothesis, we engineered the NIS mutants p.R130A and Q and functionally tested these proteins in HEK-293T cells. In steady-state iodide uptake assays, p.R130A NIS did not exhibit any significant iodide transport. However, the mutant p.R130Q displayed perchlorate-sensitive iodide transport, although to a lesser extent than WT NIS (Fig. 4B). Unlike p.R130Q, where a fraction of the mutant protein is fully glycosylated (∼100 kDa, band C) and reached the plasma membrane, the mutant protein p.R130A NIS only exhibited core glycosylation (∼60 kDa, band B) and was retained in the endoplasmic reticulum (Fig. 4C, D).
Interestingly, multiple sequence alignment of NIS orthologues and SLC5A family members revealed a high conservation of arginine and glutamine residues at the position corresponding to R130 in human NIS (Fig. 4E, F). Based on these data, we conclude that the intramolecular interaction between the β carboxyl group of D369 and the guanidinium group of R130, or alternatively the polar amide group of Q130, plays a critical role for the correct folding required for NIS maturation and transport to the plasma membrane to occur. Our hypothesis is further supported by the observation that, in the experimental structure of vSGLT, the template used for the NIS homology model, D380, is putatively involved in an ionic interaction with K127, the equivalent residues to D369 and R130 in human NIS (24).
Discussion
Patients with dyshormonogenic congenital hypothyroidism caused by different pathogenic SLC5A5 gene variants have shown a substantial clinical heterogeneity. The phenotypes of the patients vary from a compensated euthyroid state to severe hypothyroidism depending on the nature of the SLC5A5 variant and dietary iodine levels (25,26). Here, we report the identification of the compound heterozygous c.970-3C>A and c.1106A>T (p.D369V) pathogenic SLC5A5 gene variants in a patient with neonatal onset of hypothyroidism due to a defect in iodide transport. Recently, Stoupa et al. (27) reported the first case of a patient with fetal goitrous hypothyroidism harboring compound heterozygous p.G18R and p.R516* NIS pathogenic variants.
In contrast, we demonstrated that minimal p.V270E NIS-mediated iodide uptake enabled sufficient thyroid hormone biosynthesis to prevent cognitive impairment during childhood (28). A likely explanation for this situation is that the identified disease-causing NIS variants abolished iodide accumulation hampering thyroid hormonogenesis, which is compatible with the absence of 131I-iodide uptake in the thyroid, as revealed by thyroid scintigraphy.
Disruption of pre-mRNA splicing has been implicated in the etiology of numerous congenital human disorders (29). Pathogenic variants frequently affect consensus splice donor or acceptor sites, and regulatory sequences causing aberrant pre-mRNA splicing. Several reports support splicing defects as a disease-causing mechanism in congenital hypothyroidism (30 –36). Particularly, the variant c.1593C>G (also named as c.1940C>G) in the SLC5A5 gene generates the nonsense variant p.Y531* NIS as well as, a new 3′ splice acceptor site in exon 13 leading to the mis-splicing-associated nonsense variant p.S509Rfs*6 NIS (37).
Here, we provide functional evidence that the variant c.970-3C>A disrupts the canonical splice acceptor site located in intron 7 impairing the inclusion of exon 8 during NIS mRNA splicing, thus leading the nonsense variant p.Y324Hfs*148 NIS. The functional consequence of the premature stop codon might be a degradation of the mis-spliced mRNA via the nonsense-mediated mRNA decay pathway, as revealed by the algorithm NMDEscPredictor (22). Current knowledge supports that a variant should be considered pathogenic on the basis of convincing evidence supporting that it causes a premature stop codon or an in-frame deletion disrupting a functional domain (38). The absence of NIS mRNA harboring premature termination codons has been described in the thyroid tissue of patients carrying the pathogenic NIS variants (27,37).
Detailed molecular characterization of pathogenic NIS variants revealed that most of the mutant proteins are intracellularly retained (5). Here, we identified the pathogenic variant p.D369V that causes NIS retention in the endoplasmic reticulum, impairing its transport to the plasma membrane. Mechanistically, we proposed that p.D369V impairs an intramolecular ionic interaction with R130 critical for the correct folding required for NIS to export the endoplasmic reticulum. Mutations affecting the TMS9, as p.D369V does, cause defective iodide uptake, as previously reported in patients harboring the pathogenic variants p.Y348D and p.T354P (39 –41). The TMS9 contains several β-OH group-containing amino acids—that is, S353 and T354—that participate in sodium binding and translocation (39,42).
Interestingly, by investigating the rat NIS mutant p.D369A, De la Vieja et al. (39) revealed that D369 is also critical for the sodium pathway. Similarly, mutations affecting TMS9 in other members of the SLC5A family of membrane transporters, such as the high-affinity sodium/glucose cotransporter SGLT1 and the low-affinity sodium/glucose cotransporter SGLT2, also affect protein function. The SGLT1 pathogenic variants p.A388V and p.F405S found in the compound heterozygous state cause glucose-galactose malabsorption (43), and the SGLT2 pathogenic variants p.M382T and p.L387M were associated with familial renal glucosuria (44).
Sufficient dietary iodide levels and adequate NIS-mediated iodide accumulation are required to maintain a euthyroid state (45). Thus, the proposita developed dyshormonogenic congenital hypothyroidism due to a defect in iodide accumulation as result of biallelic loss-of-function NIS variants. The variant c.970-3C>A affects the splice acceptor consensus sequence in intron 7 leading to exon 8 skipping during NIS mRNA splicing. The resulting nonsense variant p.Y324Hfs*148 must be expressed at very low levels, if at all, since the corresponding mis-spliced mRNA is predicted to undergo nonsense-mediated decay. Therefore, the patient is functionally hemizygous for the pathogenic variant p.D369V identified on the other allele, which impairs NIS transport to the plasma membrane, and consequently, NIS-mediated iodide accumulation required for normal thyroid hormonogenesis.
Footnotes
Acknowledgments
We are grateful to Dr. Ari J. Wassner (Boston Children's Hospital Thyroid Center) for critical reading of the article and helpful suggestions. We thank Dr. Nancy Carrasco (Vanderbilt University School of Medicine) and Dr. Thomas v. O. Hansen (Copenhagen University Hospital, Denmark) for kindly providing critical reagents. We also thank Dr. Pilar Crespo (Centro de Micro y Nanoscopía de Córdoba, Consejo Nacional de Investigaciones Científicas y Técnicas, Universidad Nacional de Córdoba) for imaging technical assistance.
Authors' Contributions
C.E.B.B. and M.M. performed experimental work and analyzed the data. R.C.G. and V.P. provided critical technical support. P.P. and A.E.C. diagnosed and enrolled the patient in the study. A.M.M.-R., A.E.C., and J.P.N. conceived the project. J.P.N. supervised the study and drafted the article. All authors participated in writing the article and approved the final version.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Fondo para la Investigación Científica y Tecnológica–Agencia Nacional de Promoción Científica y Tecnológica (Grant Nos. PICT-2018-1596 and PICT-2019-1772) and Secretaría de Ciencia y Tecnología–Universidad Nacional de Córdoba (Grant No. 33620180100772CB).