
Abstract
We report a patient with congenital hypothyroidism due to athyreosis complicated by a heterozygous thyroid hormone receptor beta (THRβ) gene mutation (R320L), resulting in a severe resistance to thyroid hormone beta phenotype. The proband inherited the mutant allele from his father, presenting a very mild phenotype. While the precise reason for this discrepancy remains unknown, we postulate the possibility of de novo mutation and mosaicism in the father. Correlating thyrotropin (TSH) with free thyroxine (fT4) allowed us to predict the amount of fT4 required to normalize the proband's TSH, which supported the treatment with high dose of levothyroxine.
Introduction
Resistance to thyroid hormone beta (RTHβ) is a syndrome of reduced tissue responsiveness to thyroid hormone. The hallmark of RTHβ is high circulating thyroid hormone levels with unsuppressed thyrotropin (TSH). The coexistence of RTHβ and thyroid dysgenesis has been reported in 5 cases, all due to thyroid ectopy, with thyroid hormone receptor beta (THRβ) gene mutation confirmed in 4 [summarized in Ref. (1)]. Herein, we describe the occurrence of true athyreosis in a newborn with RTHβ due to a mutation in the THRβ gene. Interestingly, the father harbored the same mutation but very mild central resistance phenotype, suggesting mosaicism.
Case Report
The proband was a 5-month-old boy born to unrelated parents at 39 weeks through C-section because of breech presentation. Transferred to NICU because of respiratory distress, he remained there for 12 days for treatment of right pneumothorax managed by a chest tube. Newborn screen at 24 hours showed a TSH of 379 mU/L. A serum sample at 4 days of age documented a TSH of 135 mU/L and free thyroxine (fT4) <0.4 ng/dL. Treatment with levothyroxine (LT4) 44 μg/day (14 μg/kg) was started on day 4 of life then increased progressively in an effort to normalize the TSH. At 5 months, 112 μg/day of LT4 (16 μg/kg) reduced his TSH to 62 mU/L while total T4 (TT4) and fT4 were 15.1 μg/dL and 2.44 ng/dL, respectively.
Correlation of serum TSH with fT4 values in the course of treatment predicted that TSH would normalize when fT4 reaches a value of 4.7 ng/dL (Fig. 1A). Resistance to thyroid hormone beta (RTHβ) was suspected, leading to further investigations into the etiology of the hypothyroidism. There were no detectable human antimouse antibodies. He had several additional health problems, including positional plagiocephaly, a tethered cord that was repaired neurosurgically, and mild left sensorineural hearing loss. Head circumference grew normally at 25th centile. Growth improved progressively: in length from the 3rd centile reaching the 50th centile at 8 months and in weight from the 10th centile to the 25th over the same period. Developmental milestones remained appropriate for age.
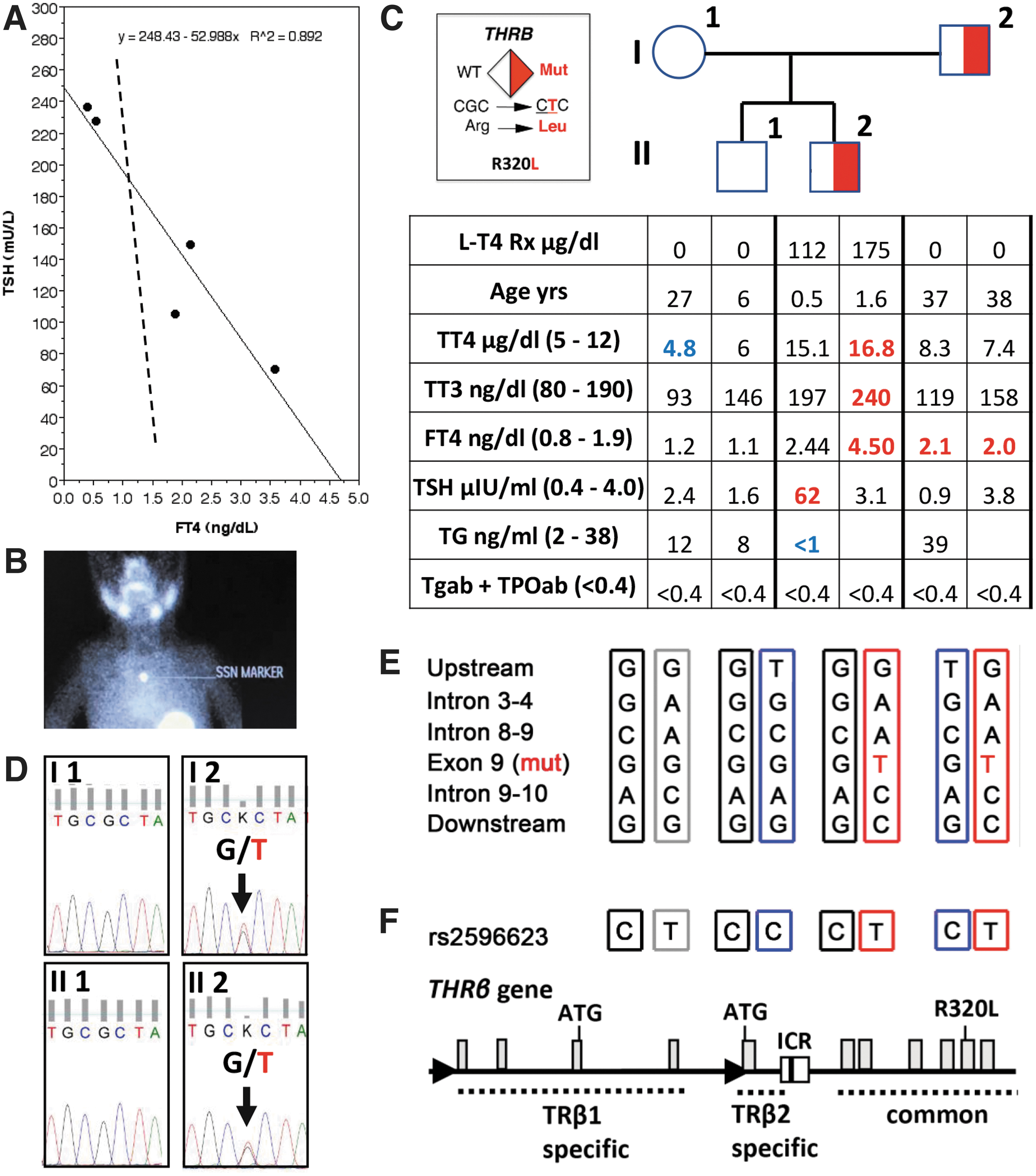
Clinical, biochemical, and genetic data on the proband and his family. (
Further Investigations
After obtaining approval from the University of Chicago Research Ethics Board and obtaining informed signed patient consents, blood was obtained from the proband and his family members. Undetectable serum thyroglobulin with high TSH suggested athyreosis, confirmed by an absent thyroidal pertechnetate uptake at 7 months of age with TSH of 29 mU/L (Fig. 1B). At 14 months of age, on 175 μg/day of LT4 (19 μg/kg), TSH was 3.1 mU/L with fT4 of 4.6 ng/dL (Fig. 1C) as predicted in Figure 1A. A heterozygous mutation in the THRβ gene (R320L; Fig. 1D) was identified.
This known mutation decreases the receptor affinity for triiodothyronine (T3) to 9.6% of the wild type (WT) (R320L Ka = 0.21 vs. WT Ka = 2.2) (2), explaining the proband's severe RTHβ. However, his father, harboring the same heterozygous THRβ mutation (Fig. 1D), had a very mild phenotype showing an fT4 of 2.1 and 2.0 ng/dL, measured 1 year apart, accompanied by normal TSH values (Fig. 1C). The mother and older brother had serum thyroid tests in the reference range and no THRβ mutation (Fig. 1C). The paternal grandmother also had normal thyroid tests and WT THRβ (data not shown).
THRβ locus haplotype showed that the proband and his brother inherited the same maternal THRβ allele, thus excluding the possibility that the maternal allele potentially contributed to the more severe phenotype in the proband (Fig. 1E). The father's distinctly mild central phenotype raises the possibility of de novo occurrence of the THRβ mutation and mosaicism as previously reported (3). Unfortunately, the paternal grandfather was not available for this study to fully assess this hypothesis. When sequencing an intronic control region (ICR) of the THRβ2 isoform (4), the proband and the father carried identical genotype (C/T in rs2596623; Fig. 1F), thus excluding the possibility that an ICR defect could lead to a differential allelic expression in their pituitary glands.
Whole-exome sequencing of the proband DNA failed to show mutations in genes known to cause thyroid dysgenesis, including thyroid transcription factors NKX2.1 and FOXE1, hematopoietically expressed homeobox protein (HHEX), and paired box 8 (PAX8). Besides, no rare or deleterious sequence abnormalities were identified in cofactors, corepressors, coactivators, and other partner proteins known to interact with the thyroid hormone receptor protein (5) that could act as phenotype modifiers responsible for the different severity of RTHβ phenotypes in the proband and father.
Discussion and Conclusion
In the 5 reported cases of RTHβ and dysgenesis, inheritance from the mother was reported in 1 case, de novo THRβ mutations in 2 cases, and the parents were not studied in the remaining two cases. A high dose of LT4 was needed to normalize the proband's TSH, confirming the prediction generated by the best fit line from early treatment data. Despite harboring the same heterozygous THRβ mutation, the proband had severe central RTHβ while the father had not. Detailed genetic investigations failed to identify additional modifiers contributing to the phenotypic discrepancy.
The proband's phenotype might have been influenced by (i) severe intrauterine hypothyroidism in setting of athyreosis and RTHβ in the fetus, limiting thyroid hormone (TH) supply to that from the mother, insufficient for a fetus with RTHβ and (ii), a delayed maturation of the feedback TSH regulation in congenital hypothyroidism, requiring higher levels of TH during early treatment (6,7). However, it is notable that the levels of fT4 relative to TSH were lower in latter setting than those observed in the current case (8, and Fig. 1A). The proband's serum fT4 was 214% above the upper limit of normal (ULN), corrected for age (9). This value is well within the respective range found in adult individuals with the same THRβ gene mutation of 170% and 200% above ULN (2).
Thus, the father's phenotype with fT4 of 108% above the ULN was incompatible not only with the proband's phenotype but also with that of previously reported patients with this mutation. Based on our previous experience with the inheritance of RTHβ by a father with mosaic of a THRβ gene mutation (3), we hypothesize that this discrepancy could be due to the same occurrence in tissues involved in central TH regulation. Unfortunately, we were unable to fully test this hypothesis as the father refused to provide additional tissue material.
To our knowledge, this represents the first report of congenital hypothyroidism due to athyreosis complicated by coincidental RTHβ.
Footnotes
Acknowledgments
We thank the family members for their willingness to participate in this study.
Authors' Contributions
F.S.-L. performed the Sanger sequencing and prepared the first draft of the article. M.M.F. analyzed the WES sequencing results. J.A.A. and J.E.W. identified the patient, instituted and supervised the treatment, and obtained the blood samples. A.M.D. and S.R. directed the study, corresponded with the treating physician, recommended the treatment, and finalized the article for review by all authors.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported, in part, by grants from The National Institutes of Health, DK15070 (to S.R.) and DK110322 (to A.D.).