
Abstract
Background:
Nonalcoholic steatohepatitis (NASH) is characterized by hepatic steatosis, lobular inflammation, and fibrosis. Thyroid hormone (TH) reduces steatosis; however, the therapeutic effect of TH on NASH-associated inflammation and fibrosis is not known. This study examined the therapeutic effect of TH on hepatic inflammation and fibrosis during NASH and investigated THs molecular actions on autophagy and mitochondrial biogenesis.
Methods:
HepG2-TRβ cells were treated with bovine serum albumin-conjugated palmitic acid (PA) to mimic lipotoxic conditions in vitro. Mice with NASH were established by feeding C57BL/6J mice Western diet with 15% fructose in drinking water for 16 weeks. These mice were administered triiodothyronine (T3)/thyroxine (T4) supplemented in drinking water for the next eight weeks.
Results:
In cultured HepG2-TRβ cells, TH treatment increased mitochondrial respiration and fatty acid oxidation under basal and PA-treated conditions, as well as decreased lipopolysaccharides and PA-stimulated inflammatory and fibrotic responses. In a dietary mouse model of NASH, TH administration decreased hepatic triglyceride content (3.19 ± 0.68 vs. 8.04 ± 0.42 mM/g liver) and hydroxyproline (1.44 ± 0.07 vs. 2.58 ± 0.30 mg/g liver) when compared with mice with untreated NASH. Metabolomics profiling of lipid metabolites showed that mice with NASH had increased triacylglycerol, diacylglycerol, monoacylglycerol, and hepatic cholesterol esters species, and these lipid species were decreased by TH treatment. Mice with NASH also showed decreased autophagic degradation as evidenced by decreased transcription Factor EB and lysosomal protease expression, and accumulation of LC3B-II and p62. TH treatment restored the level of lysosomal proteins and resolved the accumulation of LC3B-II and p62. Impaired mitochondrial biogenesis was also restored by TH. The simultaneous restoration of autophagy and mitochondrial biogenesis by TH increased β-oxidation of fatty acids. Additionally, the elevated oxidative stress and inflammasome activation in NASH liver were also decreased by TH.
Conclusions:
In a mouse model of NASH, TH restored autophagy and mitochondrial biogenesis to increase β-oxidation of fatty acids and to reduce lipotoxicity, oxidative stress, hepatic inflammation, and fibrosis. Activating thyroid hormone receptor in the liver may represent an effective strategy for NASH treatment.
Introduction
Nonalcoholic steatohepatitis (NASH) is a severe consequence of metabolic dysfunction-associated fatty liver disease (MAFLD) (1,2) and is characterized by steatosis, inflammation, hepatocyte injury, and fibrosis (3). The prevalence of NASH worldwide ranges between 1.5% and 6.45% (4), and NASH is a major cause for liver transplantation in Western countries (5). Currently, there is no FDA-approved drug for the treatment of NASH. Interestingly, patients with MAFLD have increased prevalence of overt and subclinical hypothyroidism (6,7), and patients with overt and subclinical hypothyroidism have increased prevalence of MAFLD (8,9). Furthermore, low serum free triiodothyronine and decreased thyroid function are significantly associated with NASH and advanced fibrosis (7,10,11). Recent studies also suggested that NASH may be associated with decreased hepatic deiodinase expression/activity and decreased intrahepatic triiodothyronine (T3) concentration (12,13).
In cultured hepatic cells, thyroid hormone (TH) stimulated hepatic lipophagy (14), and promoted mitochondrial biogenesis and mitophagy, to increase fatty acid β-oxidation (15,16). In dietary or genetic rodent models of hepatosteatosis, TH and thyroid hormone receptor beta (TRβ) agonists decreased hepatic triglyceride content (17 –20). Recent clinical trials showed that low-dose levothyroxine supplementation or TRβ-selective agonist decreased hepatic steatosis in patients with NAFLD (21,22). However, little is known about TH or thyromimetics on inflammation and fibrosis in NASH. Accordingly, we treated mice with NASH and with TH for eight weeks and found that TH not only reduced lipotoxic lipid species in the liver but also reduced hepatic inflammation and fibrosis.
Materials and Methods
Please see more detailed methods in Supplementary Data.
Animal models
The use of animal was approved by the Institutional Animal Care and Use Committee (IACUC) at SingHealth (2015/SHS/1104).
Diet-induced NASH mice were fed with RD Western Diet (D12079B; Research Diets, Inc.) supplemented with 15% fructose in drinking water (WDF), as previously described (23). Ten-week-old male C57BL/6J mice were fed with normal chow diet (NCD) or WDF for 16 weeks, at which point half of the WDF-fed mice began receiving T3 (35 ng/g mice) and thyroxine (T4) (7 ng/g mice) in 15% (w/v) fructose drinking water for the next 8 weeks (24). Body weight was monitored weekly. Drinking water was replaced weekly, and water intake was documented by measuring the volume before and after replacement.
The T3 concentration (ng/mL) was calculated by
Cell culture
HepG2 transformants expressing ectopic TRβ1 (HepG2-TRβ1) were generated, as previously described (25). HepG2-TRβ1 cells were maintained at 37°C in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum in a 5% CO2 atmosphere.
To investigate the effect of TH on mitochondrial activity, HepG2-TRβ cells were treated with or without 1, 10, or 100 nM T3 for 24 hours, followed by co-treatment with 0.5% bovine serum albumin-conjugated palmitic acid (PA) (0.5 mM) for another 24 hours.
To investigate the effect of T3 on lipopolysaccharides (LPS) and PA-induced inflammatory responses, HepG2-TRβ cells were treated with or without T3 (100 nM) in the presence or absence of LPS (100 ng/mL) and PA (0.2 mM) for 24 hours.
Cellular oxygen consumption rate analysis
Oxygen consumption rate (OCR) was measured using an XFe96 Extracellular Flux Analyzer (Seahorse Bioscience, Inc.). Oligomycin (1 μM), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (1 μM), rotenone (1 μM) and antimycin A (1 μM), and etomoxir (4 μM) were added according to manufacturer's instructions.
Statistical analyses
The comparison between NASH mice versus control mice, and TH-treated NASH mice versus NASH mice, was analyzed using unpaired parametric Student's t-test (two-tailed). The comparison between control, NASH, and TH-treated NASH mice was conducted using ordinary one-way analysis of variance, followed by multiple comparisons using Fisher's least significant difference test. Heat map of metabolites was presented as mean value of each group using normalized percentage value for each lipid species, with smallest mean defined as 0% and largest mean defined as 100%. All the analyses were performed using GraphPad Prism 8.
Results
TH increased fatty acid oxidation and decreased lipotoxicity in cultured hepatic cells
To determine whether TH could potentially be effective in reducing the inflammation and fibrosis of NASH, we first examined THs effects on mitochondrial function and the expression of inflammation and fibrosis genes in cultured hepatic cells under lipotoxic conditions. We treated TRβ-expressing HepG2 (HepG2-TRβ) cells with PA, a saturated fatty acid that causes lipotoxicity and impairs mitochondrial function (26), and analyzed the effects of TH on OCR. The drop in OCR after addition of ATP synthase inhibitor oligomycin (Oligo) reflects ATP production. Addition of mitochondrial uncoupling agent FCCP drives mitochondrial respiration to a maximal level. The OCR after addition of complex I and III inhibitors rotenone and antimycin A (R+A) reflects non-mitochondrial respiration. We found that PA decreased basal respiration, ATP production, and coupling efficiency compared with untreated control. In contrast, TH increased basal and maximum respiration, spare respiratory capacity, ATP production, and coupling efficiency to similar levels in cells cultured with or without PA (Fig. 1A and Supplementary Fig. S1A).

TH increased mitochondrial lipid oxidation and reduced inflammatory response in cultured HepG2-TRβ cells. (
We next determined the effects of TH on mitochondrial fatty acid β-oxidation by treating cells with the CPT1α inhibitor, etomoxir. The resultant decrease in OCR after etomoxir addition is an indicator of mitochondrial fatty acid β-oxidation. We found that PA caused less reduction in OCR than control cells, consistent with PA decreasing β-oxidation of fatty acids. TH caused a more pronounced reduction in OCR in cells treated with or without PA (Fig. 1B and Supplementary Fig. S1B), indicating that TH increased fatty acid oxidation during both conditions. Furthermore, PA and LPS co-treatment stimulated the expression of inflammatory and fibrotic genes (Fig. 1C). T3 abrogated the induction of these genes (Fig. 1C). Taken together, these results suggested that T3 promoted mitochondrial function and lipid oxidation and reduced inflammatory and fibrotic response in cultured HepG2-TRβ cells during lipotoxic conditions.
TH decreased steatosis in mice with NASH
Previous studies showed that mice fed a Western diet supplemented with liquid fructose (WDF) developed NASH (23). We thus characterized mice fed WDF or NCD for 16 weeks and found that they had increased circulating ALT, hepatic triglyceride content, hepatic inflammation, and fibrosis (Supplementary Fig. S2). These results demonstrated that mice developed NASH after being fed WDF for 16 weeks.
To determine the therapeutic effect of TH on NASH, we first fed mice WDF for 16 weeks and then administrated TH in drinking water (T3: 35 ng/g mice, T4: 7 ng/g mice) (24) from weeks 17 to 24 (TH-treated NASH mice, Fig. 2A), and compared them with mice fed continuously with NCD or WDF for 24 weeks. This chronic oral TH regimen previously was shown to cause approximately threefold increase of circulating T3 and 80% reduction of T4 (24), consistent with mild hyperthyroidism in mice. To verify TH action on hepatic cells, we measured gene expression of Dio1, a well-characterized functional marker of hepatic TH level, and found that Dio1 mRNA expression was increased in NASH mice treated with TH (Supplementary Fig. S3).
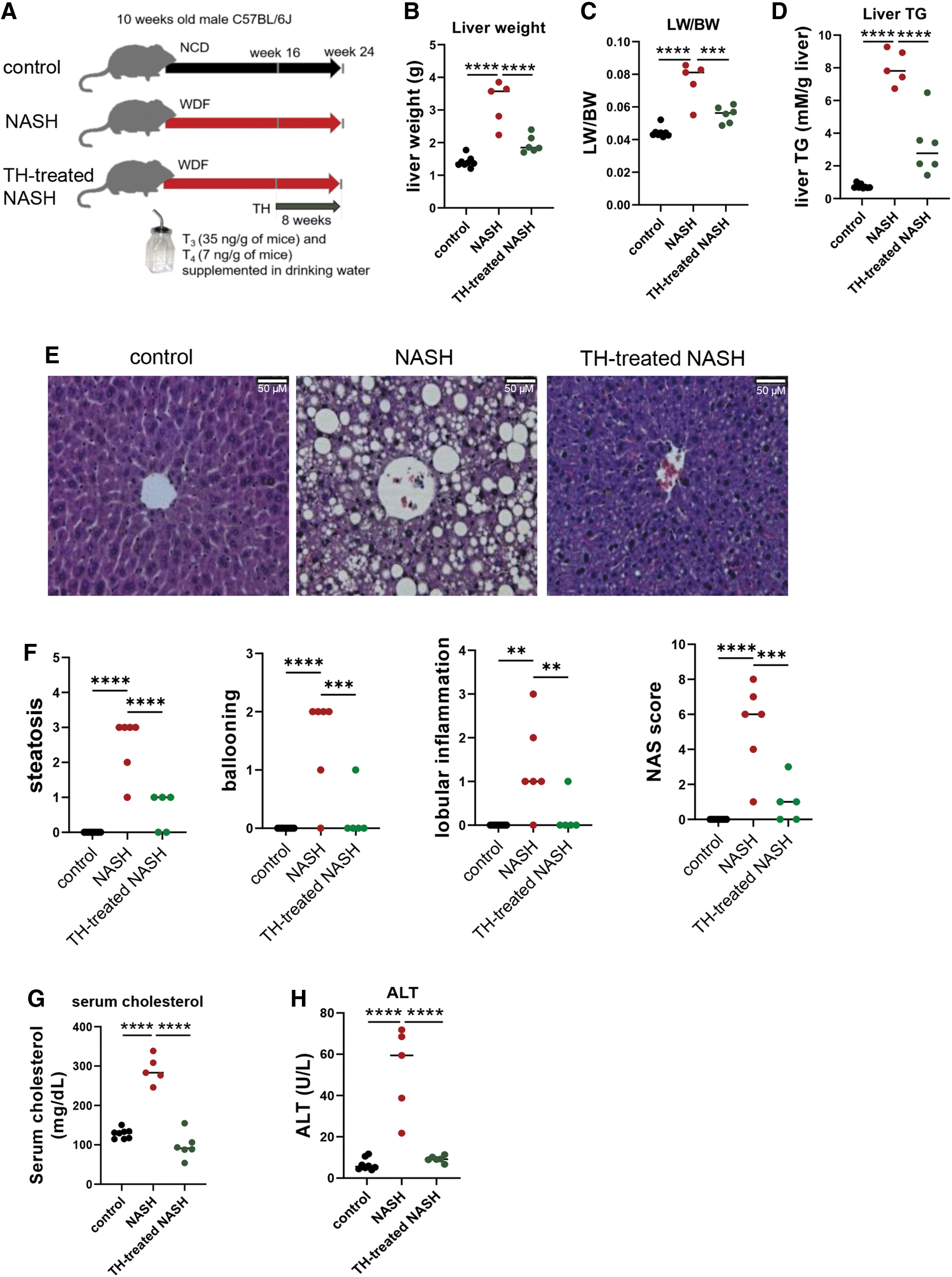
TH decreased hepatic steatosis as well as serum cholesterol and ALT in NASH mice. (
TH-treated NASH mice had decreased liver weight (1.9 ± 0.1 vs. 3.2 ± 0.3 g), liver index (liver weight to body weight ratio; LW/BW), and hepatic triglyceride content (3.2 ± 0.7 vs. 8.0 ± 0.4 mM/g liver) compared with NASH mice (Fig. 2B–D). Hematoxylin–eosin staining also showed decreased hepatic lipid content in TH-treated NASH mice (Fig. 2E). When compared with mice with NASH, TH-treated NASH mice had decreased steatosis (0.6 ± 0.2 vs. 2.5 ± 0.3), ballooning (0.2 ± 0.2 vs. 1.5 ± 0.3), lobular inflammation (0.2 ± 0.2 vs. 1.3 ± 0.4), and NAFLD activity score (NAS) (1 ± 0.5 vs. 5.3 ± 0.9) (Fig. 2F). Additionally, TH-treated NASH mice had decreased levels of serum cholesterol (97.4 ± 12.3 vs. 290.6 ± 14.0 mg/dL) and ALT (9.2 ± 0.6 vs. 52.0 ± 8.5 U/L) compared with NASH mice (Fig. 2G, H). Taken together, our data demonstrated that TH decreased hepatic steatosis and liver damage in NASH liver.
TH decreased lipotoxic lipids in NASH liver
We next performed pathway analysis of the hepatic transcriptomes of these mice. Pathways that showed the most statistically significant upregulation in NASH mice versus control mice were inflammation, chemotaxis, and extracellular matrix organization (Supplementary Fig. S4A). Similar pathways were downregulated in TH-treated NASH mice versus NASH mice (Supplementary Fig. S4B). Interestingly, the main metabolic pathways upregulated by TH administration were hepatic organic acid, cholesterol, carbohydrate, and fatty acid metabolic processes (Supplementary Fig. S4B). These findings suggested that TH might exert its beneficial effects during NASH by modulating hepatic lipid metabolism.
We next performed lipidomic profiling of triacylglycerols (TAGs), diacylglycerols (DAGs), monoacylglycerols (MAGs), and cholesterol esters (CEs) and found that these lipid metabolites accumulated in livers of mice with NASH. Remarkably, TH treatment decreased these lipid species to near-baseline levels (Fig. 3A–D), as well as decreased total hepatic TAGs (295.3 ± 99.9 vs. 1144.4 ± 542.8 μM/g liver), DAGs (0.015 ± 0.004 vs. 0.053 ± 0.024 μM/g liver), MAGs (0.007 ± 0.002 vs. 0.023 ± 0.01 μM/g liver), and CEs (8.02 ± 3.45 vs. 26.49 ± 8.38 μM/g liver) compared with untreated NASH mice (Fig. 3E).
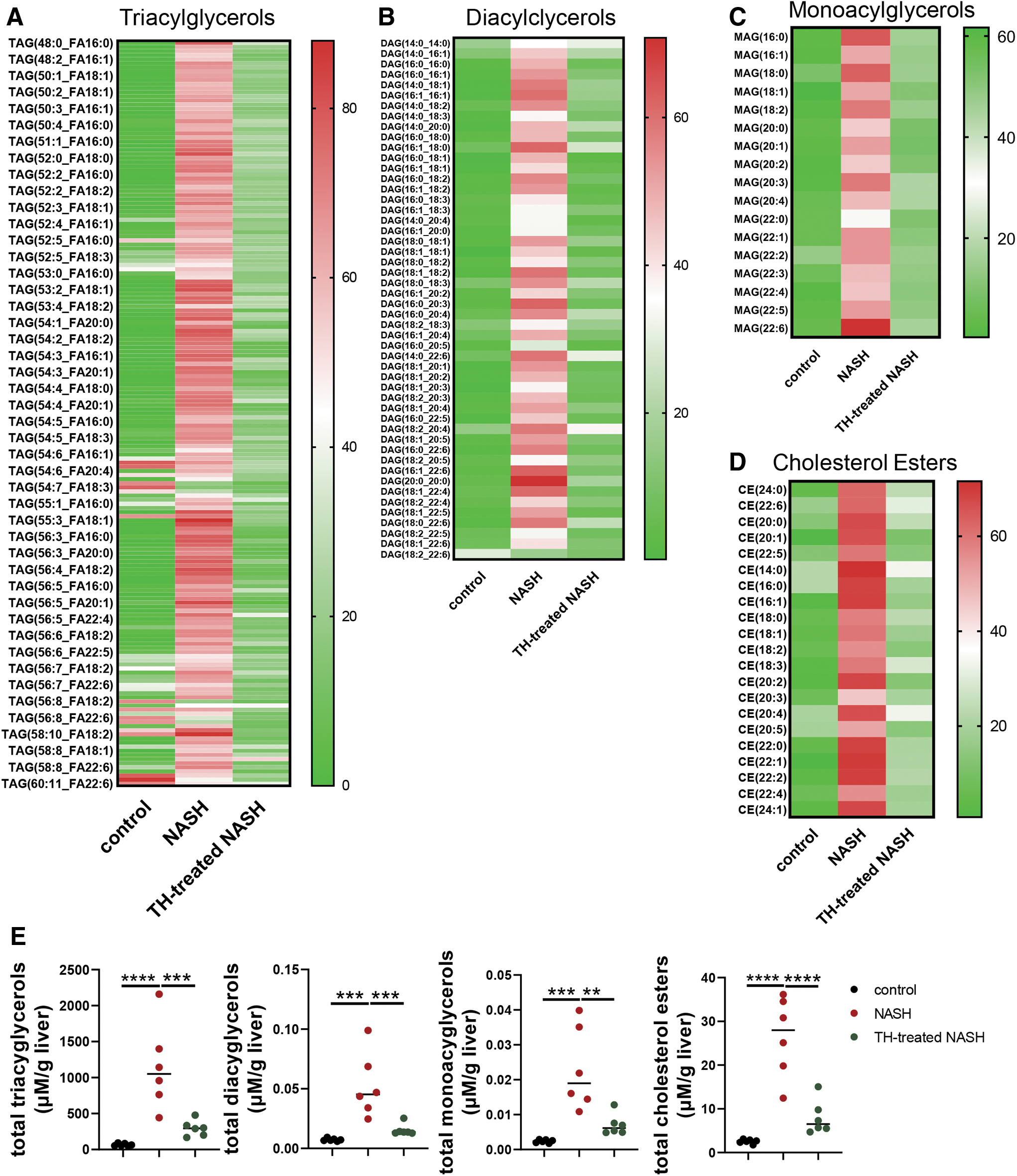
TH-treated NASH mice displayed improved hepatic lipidomic profile. Relative hepatic TAG (
TH increased hepatic autophagy to promote β-oxidation of fatty acids in mice with NASH
We next investigated whether the decrease in hepatic acylglycerols could be attributed to increased β-oxidation of fatty acids by TH by measuring the serum levels of β-hydroxybutyrate, the end product of β-oxidation of fatty acids. There were no significant differences in serum β-hydroxybutyrate levels between control and NASH mice; however, TH-treated mice had increased serum levels of β-hydroxybutyrate compared with NASH mice (0.55 ± 0.07 vs. 0.21 ± 0.01 mM) (Fig. 4A). These findings showed that TH increased β-oxidation of fatty acids in mice with NASH.

TH increased hepatic autophagy to enhance β-oxidation of fatty acids in NASH mice. (
We previously demonstrated that TH induction of β-oxidation required autophagy–lysosomal breakdown of triglycerides stored in fat droplets (lipophagy) (14). Accordingly, we investigated autophagy in control and NASH mice and found that both LC3B-II and p62 levels were increased in NASH mice (Fig. 4B), whereas the mRNA levels of LC3B-II was unchanged, and mRNA level of p62 was decreased (Supplementary Fig. S5). Since both p62 and LC3B-II are normally degraded, and accumulate when autophagic flux is inhibited (27), these findings indicated that hepatic autophagic degradation was decreased in NASH.
In support of this notion, we found the protein levels of lysosomal protease, cathepsin D, and the key transcription factor for lysosomal biogenesis, transcription Factor EB (TFEB), were downregulated in NASH mice compared with control mice (Fig. 4B). In contrast, there was increased TFEB, cathepsin D, and lysosomal protein LAMP2, and decreased protein levels of p62 and LC3B-II in livers of TH-treated NASH mice than untreated NASH mice (Fig. 4B), indicating that TH treatment restored lysosomal function and autophagic degradation. Using transmission electron microscopy (TEM), we found that TH-treated NASH mice displayed lipid-laden autolysosomes (Fig. 4C), confirming that TH increased lipophagy to release free fatty acids for β-oxidation.
We also quantified mitochondrial number in hepatocytes by TEM and found that it was decreased in NASH mice, whereas it was increased in TH-treated NASH mice (Fig. 4D and Supplementary Fig. S6). Profiling of acylcarnitines showed that long- and medium-chain acylcarnitines were increased in livers of TH-treated mice with NASH (Fig. 4E), indicating that TH increased hepatic acylcarnitine flux and β-oxidation of long-chain fatty acids in these mice. Altogether, our data showed that TH restored hepatic autophagic flux and increased lipophagy to increase mitochondrial β-oxidation of fatty acids in mice with NASH.
Interestingly, TH also increased mRNA expression of lipases, Atgl and Mgll (Supplementary Fig. S7A), and decreased mRNA expression of several lipogenesis genes, including SREBP1c (encoded by Srebf1 gene), Thrsp, Acc, and Scd1. TH did not change the mRNA expressions of Fasn, Dgat2, and chREBP (Supplementary Fig. S7B). Taken together, our data suggested that TH also decreased lipogenesis and increased lipolysis in the liver during NASH.
TH increased mitochondrial content to promote β-oxidation of fatty acids in mice with NASH
We next determined mitochondrial content by measuring hepatic mitochondrial DNA copy number and found that it was decreased in NASH mice and was restored to baseline levels by TH (Fig. 5A). The hepatic expression of mitochondrial proteins, COXIV and VDAC1, was also decreased in NASH mice and was restored in TH-treated NASH mice (Fig. 5B). Protein level of PGC1α, an important cofactor for mitochondrial biogenesis, was upregulated by TH (Supplementary Fig. S8). Taken together with our earlier TEM studies showing that TH increased lipophagy and hepatic mitochondrial number in mice with NASH (Fig. 4D), these results showed that TH simultaneously restored autophagy and mitochondrial biogenesis to increase β-oxidation in NASH liver.
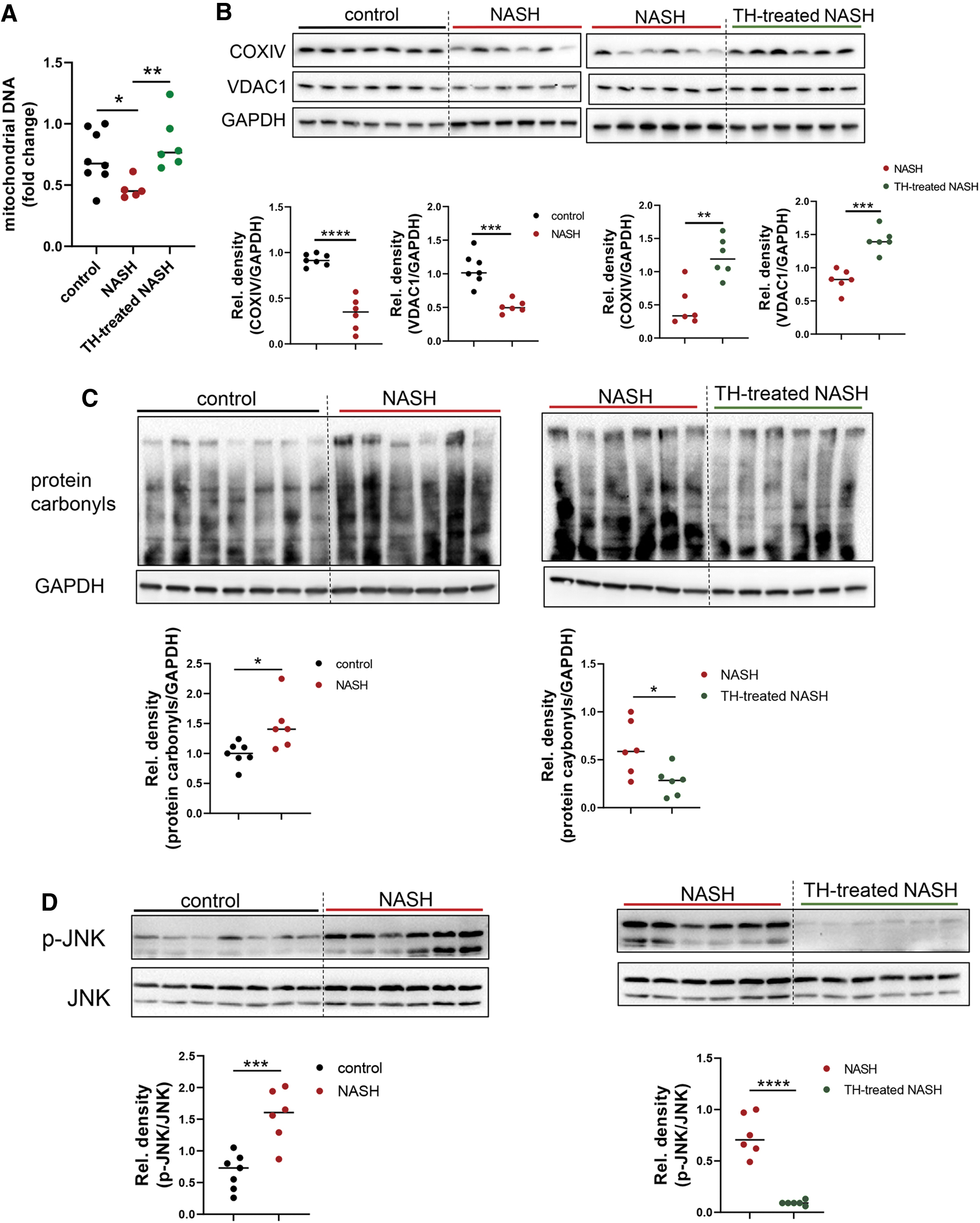
TH-treated NASH mice showed increased hepatic mitochondrial content and function. (
TH decreased hepatic oxidative stress and inflammasome activation in mice with NASH
Increased lipotoxicity is associated with increased oxidative stress, activation of JNK signaling, and NLRP3 inflammasome formation (28). We thus examined hepatic oxidative stress by detecting carbonyl groups introduced into proteins by oxidative reactions and found that protein carbonyls were increased in NASH mice compared with control mice. In contrast, TH-treated NASH mice decreased hepatic protein carbonyls to levels similar to control mice (Fig. 5C). Similarly, TH treatment abolished the increased p-JNK in NASH livers (Fig. 5D).
Several factors associated with hepatic steatosis/inflammation contributed to the activation of NLRP3 inflammasome, including LPS, TNFα, IL-1β, and oxidative stress (29 –31). NLRP3 activation in NASH led to severe hepatic inflammation and fibrosis (31). In this connection, NLRP3 inflammasome components NLRP3, ASC1, and caspase 1, and toll-like receptor 4 (TLR4) mRNA and protein expressions were increased in livers of NASH mice (Fig. 6) and markedly decreased by TH (Fig. 6). TH can suppress the expression of NLRP3 components during ischemia–reperfusion in an AMPK-dependent manner (32). We also found that TH treatment increased AMPK phosphorylation in mice with NASH (Supplementary Fig. S9). Taken together, our data demonstrated that TH treatment reduced lipotoxicity-associated oxidative stress and NLRP3 inflammasome activation in NASH liver.
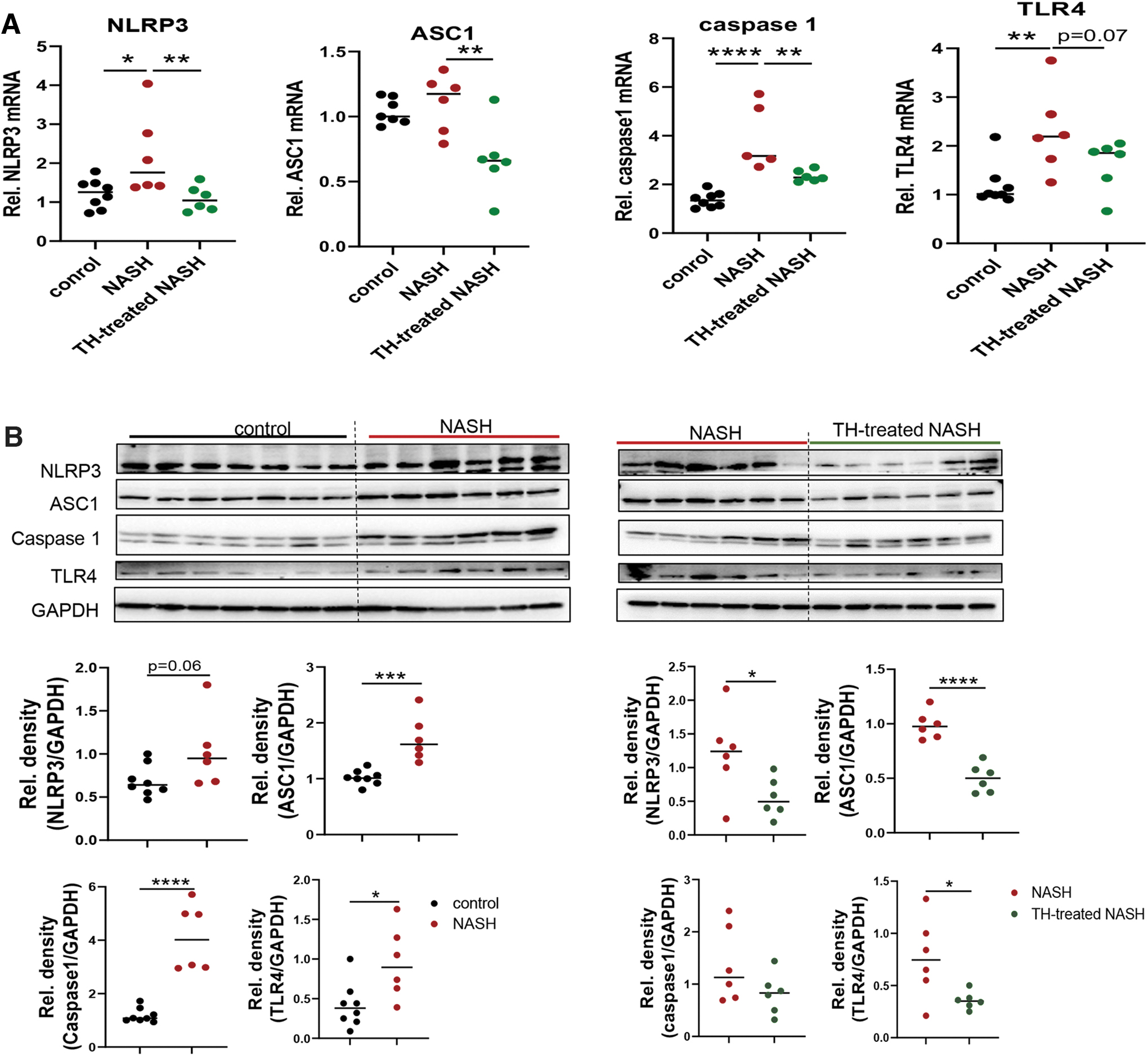
TH-treated NASH mice showed decreased NLRP3 inflammasome activation in the liver. (
TH decreased inflammatory cytokines, chemokines, and collagen production in livers of mice with NASH
Hepatic mRNA expressions of cytokines and chemokines were upregulated, and their inductions were reduced by TH treatment in mice with NASH (Fig. 7A). They also had increased hepatic hydroxyproline content compared with control mice. In contrast, TH treatment decreased hydroxyproline content of mice with NASH (1.44 ± 0.07 vs. 2.6 ± 0.3 mg/g liver) (Fig. 7B). Decreased hepatic fibrosis by TH was further confirmed using Masson's trichrome staining (Fig. 7C). The hepatic mRNA levels of fibrotic markers including TGFβ (encoded by Tgfb gene) and several collagen genes were elevated in NASH mice but reduced by TH (Fig. 7D). Taken together, these results showed that TH treatment decreased hepatic inflammation and fibrosis in mice with NASH.
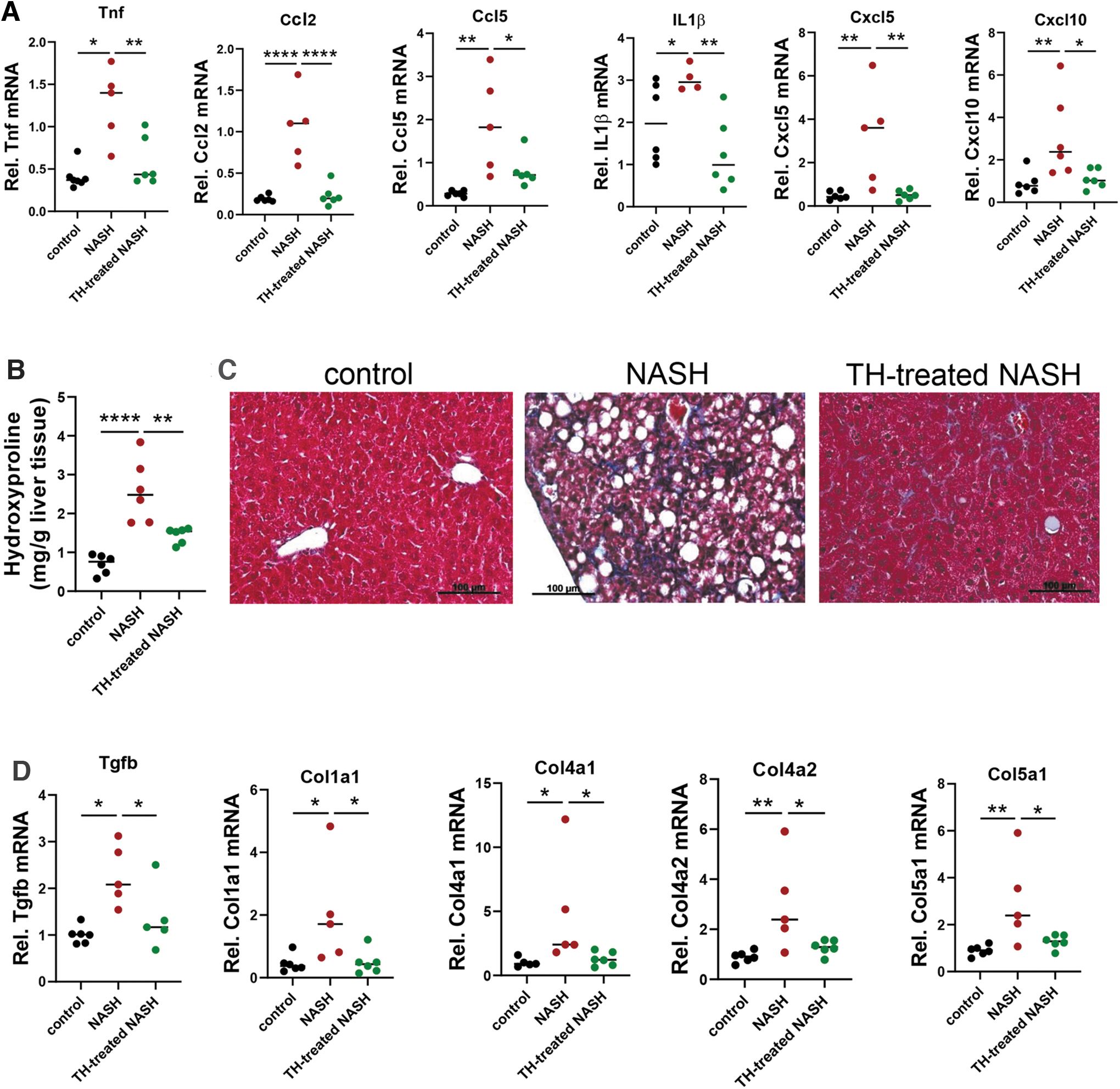
TH-treated NASH mice showed decreased hepatic inflammation and fibrosis. (
Discussion
Although several studies previously demonstrated the effectiveness of TH in reducing hepatic steatosis in animal models (12), the effectiveness of TH on hepatic inflammation and fibrosis in NASH remains unknown. In our current study, we first induced NASH in mice by feeding them with WDF for 16 weeks and then treated them with TH for 8 weeks to assess the therapeutic effect of TH in pre-existing NASH (Fig. 2). We found that TH decreased lipotoxic lipid species in the liver (Fig. 3), reduced oxidative stress and NLRP3 inflammasome activation (Figs. 5 and 6), and hepatic inflammation, NAS score, and hepatic fibrosis in mice with NASH (Figs. 2, 6, and 7).
Autophagy is a major mechanism for mobilizing triglycerides from lipid droplets and releasing free fatty acids for mitochondrial β-oxidation (14,16,33). We previously demonstrated that TH injection for 3 days to mice fed NCD or TH injection for 7 days to mice fed high fat diet led to increased hepatic autophagy and fatty acid β-oxidation (14,34). In contrast, in patients with NAFLD, autophagy was impaired as evidenced by decreased lysosomal proteases and accumulation of both LC3B-II and p62 (35,36). We also found that mice with NASH faithfully recapitulated the impairment of autophagy that was observed in patients with NASH (Fig. 4B). TH treatment restored the levels of lysosomal proteins and autophagy degradation in NASH livers as evidenced by the reversal of LC3B-II and p62 accumulation (Fig. 4B).
TH also increased lysosomal mobilization of lipid droplets (Fig. 4C), import of fatty acids into the mitochondria as acylcarnitines (Fig. 4E), and increased serum levels of β-hydroxybutyrate (Fig. 4A), consistent with increased β-oxidation. Mice with NASH also showed impaired mitochondrial number, which also was restored by TH treatment (Figs. 4D and 5A, B). Thus, in mice with NASH, TH treatment restored both autophagy and mitochondrial function to increase fatty acid β-oxidation and reduced lipotoxic lipid species and stress response. Additionally, TH induction of autophagy may have had a suppressive effect on inflammasome formation and thus represents another mechanism for TH-mediated reduction of hepatic inflammation and fibrosis since autophagy can reduce inflammasome formation (37).
There remain legitimate medical concerns about the adverse effects of TH on the heart and bone when considering the therapeutic use of TH for NASH. However, 16-week supplementation of low-dose levothyroxine with careful titration in diabetic male patients reduced intrahepatic lipid content without causing significant adverse effects in almost all patients (21), suggesting that short-term therapy may be safe and effective. We used a combination of T3 and T4 in these studies, so it remains to be determined whether T4, which is less potent but has less side effects than T3, can have beneficial effects on NASH when used alone.
The natural ligands, T4 and T3, were used in this study, so the beneficial effects by TH were likely mediated by both TRα and TRβ isoforms present in the liver. Previous studies showed that TH induced similar hepatic transcriptome profiles in TRα and TRβ knockout mice (38). However, since TRβ is the dominant isoform and represents 80% of TRs in the liver (39), thyromimetics that selectively target TRβ or the liver have been developed to treat hypercholesterolemia and NASH to minimize the potential side effects of TH on other tissues that predominantly express TRα (18). The TRβ-selective thyromimetic, resmetirom (MGL-3196), and liver-specific thyromimetic, VK2809, currently are undergoing phase 3 (NCT03900429) and phase 2b clinical trials (NCT04173065) for NASH. Our study serves as a proof-of-principle by demonstrating that a combination of the natural ligands for TR, T3 and T4 was effective in reducing hepatic inflammation and fibrosis in a dietary mouse model of NASH. Thus, activating TR signaling in the liver is promising strategy to treat NASH.
Footnotes
Authors’ Contributions
J.Z. and P.M.Y. conceived the experiments. J.Z., M.T., J.P.H., A.A.W., S.G.S., M.D.C., A.J., and Y.W. performed the experiments. J.C., J.-P.K., K.-H.L., S.A.C., and B.-H.B. established experimental protocol. J.Z., M.T., and A.A.W. analyzed the data. J.Z. and P.M.Y. wrote the article. All authors provided critical feedback, helped shape the research, analysis, and article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Ministry of Health, A*STAR and National Medical Research Council Singapore grants MOH-000306 (MOH-CSASI19may-0001) to P.M.Y.; NMRC/OFYIRG/077/2018 to M.T.; Duke-NUS Medical School and Estate of Tan Sri Khoo Teck Puat Khoo Pilot Award (Collaborative) Duke-NUS-KP (Coll)/2018/0007A to J.Z.
Data Availability Statement
The RNA-seq data associated with the article are available at GEO repository (GSE172297).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9