
Abstract
Background:
Monocarboxylate transporter 8 (MCT8) deficiency is a rare genetic disease leading to a severe developmental delay due to a lack of thyroid hormones (THs) during critical stages of human brain development. Some MCT8-deficient patients are not as severely affected as others. Previously, we hypothesized that these patients' mutations do not affect the functionality but destabilize the MCT8 protein, leading to a diminished number of functional MCT8 molecules at the cell surface.
Methods:
We have already demonstrated that the chemical chaperone sodium phenylbutyrate (NaPB) rescues the function of these mutants by stabilizing their protein expression in an overexpressing cell system. Here, we expanded our previous work and used iPSC (induced pluripotent stem cell)-derived brain microvascular endothelial-like cells (iBMECs) as a physiologically relevant cell model of human origin to test for NaPB responsiveness. The effects on mutant MCT8 expression and function were tested by Western blotting and radioactive uptake assays.
Results:
We found that NaPB rescues decreased mutant MCT8 expression and restores transport function in iBMECs carrying patient's mutation MCT8-P321L. Further, we identified MCT10 as an alternative TH transporter in iBMECs that contributes to triiodothyronine uptake, the biological active TH. Our results indicate an upregulation of MCT10 after NaPB treatment. In addition, we detected an increase in thyroxine (T4) uptake after NaPB treatment that was not mediated by rescued MCT8 but an unidentified T4 transporter.
Conclusions:
We demonstrate that NaPB is suitable to stabilize a pathogenic missense mutation in a human-derived cell model. Further, it activates TH transport independent of MCT8. Both options fuel future studies to investigate repurposing the Food and Drug Administration-approved drug NaPB in selected cases of MCT8 deficiency.
Introduction
Monocarboxylate transporter 8 (MCT8) deficiency (OMIM 300523) is caused by mutations in the solute carrier family 16, member 2 (SLC16A2) gene located on the X chromosome. Affected males suffer from neurocognitive impairments as well as signs of thyrotoxicosis in peripheral organs. Common symptoms of MCT8 deficiency are global muscle hypotonia and severe developmental delay, including a lack of speech development (1).
MCT8 is characterized as a thyroid hormone (TH) transporter that facilitates the transport of 3,3′,5,5′-tetraiodothyronine (thyroxine [T4]) and 3,3′,5-triiodothyronine (rT3) as well as their metabolites rT3 and 3,3′-diiodothyronine across plasma membranes (2,3).
Proper development of the brain depends on the availability of adequate amounts of TH in brain cells during the right developmental period (4). To find molecular reasons for the neurodevelopmental delay caused by MCT8 deficiency, human (fetal) brains have been investigated for spatiotemporal MCT8 expression patterns (5 –7). Among others, MCT8 showed high expression in vascular structures that supported the idea that it is important for the transport of TH across the blood–brain-barrier (BBB) in humans (7,8). Similar observations have been made earlier in rodents (9 –11).
Mct8-deficient mouse models replicate the endocrinological phenotype described in patients with high triiodothyronine (T3) levels, low T4 levels, and high to normal thyrotropin levels in plasma (9,12 –14). However, Mct8-deficient mice have not been demonstrated to suffer from cognitive impairments. Although the uptake of T3 into the brains of these animals is strongly impaired, T3 content in the brain remains normal (10).
Results from different mouse studies suggested a compensating mechanism that includes T4 import by the organic anion transporting polypeptide 1c1 (Oatp1c1) and its conversion to T3 by the enzyme deiodinase type 2 (Dio2). Oatp1c1 is a T4 (but not T3) transporter, which is expressed at the BBB during early stages of mouse brain development, but it is absent during similar developmental stages in primates and humans (11,15,16). Genetic inactivation of Dio2 in Mct8-deficient mice shows that increased conversion of T4 into T3 compensates partially for the reduced uptake of T3 (17).
Both Dio2/Mct8-deficient mice and Oatp1c1/Mct8-deficient mice exhibit impaired development of parvalbumin-expressing neurons and decreased motor skills, thus resembling more closely the phenotype of MCT8-deficient patients (17,18).
We have previously shown that the chemical chaperone sodium phenylbutyrate (NaPB) is able to rescue protein expression and increase transport function of several unstable pathogenic MCT8 mutants in transfected cells (19,20). Here, we expanded our previous work and tested whether NaPB is able to rescue pathogenic MCT8-P321L expressed in human-induced pluripotent stem cell (iPSC)-derived brain microvascular endothelial-like cells (iBMECs), which were differentiated from patient-specific cells. It was shown earlier that pathogenic P321L-iBMECs display reduced T3 and T4 uptake compared with their isogenic control (8).
We demonstrate here that the P321L variant exhibits reduced protein expression, which can be rescued by NaPB treatment, leading to a restoration of TH uptake. We further show that the related TH transporter MCT10 contributes to the uptake of T3 into iBMECs.
Our data suggest that NaPB, which is an approved drug that is already used for chronic treatment of urea cycle defects (21), could be repurposed as a possible drug candidate for the treatment of MCT8 deficiency.
Materials and Methods
Descriptions of iPS cell lines and differentiation into iBMECs
Fibroblasts from skin biopsies from an MCT8-deficient patient and his father were obtained under consent and privacy guidelines of the University of Chicago. All procedures were performed in accordance with the Institutional Review Board (IRB) guidelines at the Cedars-Sinai Medical Center under IRB-SCRO Protocols Pro00021505 and Pro00032834.
Reprogramming and characterization of MCT8-deficient iPSCs as well as their differentiation into iBMECs are described in Vatine et al. (8).
Chaperone treatment
NaPB was purchased from Santa Cruz (Dallas, TX) and dissolved in human endothelial serum-free medium (hESFM; ThermoFisher Scientific, Waltham, MA) +1% platelet-poor plasma derived bovine serum (PPBS; Bioquote, York, UK) to a stock concentration of 8 mM. Treatment of iBMECs with 2 mM NaPB diluted in medium was performed for 24 hours the day after cleaning. Higher concentrations of NaPB and longer duration of treatment led to increased cell death. Increased cell death is probably explained by the ability of NaPB to inhibit proliferation (22).
Western blotting and quantification
Pellets from iBMECs grown on 6 cm plates were lysed as described in Braun et al. (23), and Braun and Schweizer (24). If not indicated differently in the figure legends, 20 μg of whole cell homogenates were blotted as explained in Braun et al. (23), and Braun and Schweizer (24). Antibodies used are given in Table 1.
Antibodies for Western Blotting and Immunohistochemistry
AB, antibody; RRID, Research Resource Identifier; PECAM-1, platelet endothelial cell adhesion molecule-1.
Immunocytochemistry
iBMECs were seeded on collagen IV—(400 mg/mL) and fibronectin—(100 mg/mL) coated cover slips. Immunocytochemistry was performed as described in Braun and Schweizer (24). Antibodies are given in Table 1.
Fluorescein assay
Fluorescein was purchased from Sigma-Aldrich and dissolved in distilled water to a stock concentration of 50 mM. One micromolar of fluorescein was added to the upper compartment of a transwell. Samples (200 μL) from the lower compartment were transferred to a black, flat-bottom 96-well plate (Greiner, Kremsmünster, Austria) after the indicated time. The lower compartment was refilled with fresh hESFM +1% PPBS. A standard curve ranging from 1 to 125 nM fluorescein was used for the calculation. The fluorescence was measured in a TECAN infinite® 200 PRO reader (TECAN, Männedorf, Switzerland) with an excitation wavelength of 460 nm and an emission wavelength of 516 nm.
Radioactive uptake assay
Radioactive uptake assays were performed as described in Braun et al. (23), and Braun and Schweizer (24). Ten nanomolar of purified 125I-T3 or 125I-T4 were diluted in Dulbecco's modified Eagle medium (DMEM)/F12 (1:1) (GIBCO, Waltham, MA), if not indicated differently in the figure legends. Measured values as counts per minutes were normalized to protein content (23,24).
Competition and inhibition studies in BMECs
Amino acids (aa; Leu, Phe, Trp) and 2-amino-2-norbornanecarboxylic acid (BCH) were purchased from Sigma-Aldrich and dissolved in uptake buffer (for buffer composition see Braun et al. [23]) to a stock concentration of 50 and 10 mM, respectively. For competition/inhibition studies, 5 mM aa and 1 mM BCH, respectively, together with 10 nM purified radioactive tracer were diluted in uptake buffer and incubated with the cells.
Silychristin (SC) was obtained from PhytoLabs (Vestenbergsgreuth, Germany). JPH203, a selective L-type amino acid transporter 1 (LAT1) inhibitor, was purchased from Sigma-Aldrich. Both compounds were dissolved in dimethyl sulfoxide (DMSO) to a stock concentration of 40 mM. Ten micromolar SC and JPH203, respectively, were directly given to the cells together with 10 nM purified radioactive tracer diluted in DMEM/F12 (1:1) or uptake buffer. DMSO served as solvent controls.
RNA Isolation and 3′-messenger RNA sequencing
Isolation of total RNA of pellets from NaPB-treated and -untreated iBMECs is described in Braun and Schweizer (24). The Next Generation Sequencing (NGS) Core Facility of the Medical Faculty of the University of Bonn was responsible for performing 3′-messenger RNA (mRNA) sequencing. Data processing was performed as previously described (25): The QuantSeq 3′-mRNA-Seq Fw. Library Prep Kit (Lexogen, Greenland, NH) was used for library preparation. Preprocessing of the data was done as recommended by the manufacturer. We used STAR aligner 2.6.0a for alignment against the GRCh38 human genome retrieved from Ensembl with the options recommended by the manufacturer. Raw data and raw counts from 3′-mRNA sequencing may be obtained from gene expression omnibus (GEO) (GSE176188).
Ribosome profiling
Treatment of the samples was performed according to McGlincy and Ingolia (26), with the exception of renunciation of any translation inhibitor in the media as well as in the buffers. Two hundred microliters of the lysate were incubated for 45 minutes with 1000 U of RNase I (ThermoFisher Scientific) at 25°C and 1300 rpm (Eppendorf centrifuge 5430 R) in a Thermo block. Ribosome protected fragments (RPF) were pelleted as described by sucrose cushion.
After T4 Polynucleotide kinase (ThermoFisher Scientific) treatment, the libraries were prepared with the TrueQuant SmallRNA-NGS Kit (GenXPro, Frankfurt am Main, Germany) and ribosomal RNA was depleted with Ribo-Zero Gold (Illumina, San Diego, CA). Sequencing was done on an Illumina NextSeq500 machine with a depth of 30 M raw reads. Data were analyzed as previously reported (27). The RPF with sizes of 20, 21, 28, and 29 nucleotides were used with an offset of 12 nucleotides from the 5′-end to the P-site. The RPF sitting with A-sites on the coding sequences of the mRNAs were counted and used for differential analysis. Raw data and raw counts from ribosome profiling may be obtained from GEO (GSE176188).
Inhibition study with JPH203 in overexpressing Madin-Darby canine kidney cells
We used human N-terminally hemagglutinin-tagged full-length MCT8 (1–613 aa) for cloning into pcDNA3 as previously described (3). The generation of MCT8-expressing as well as mock-transfected Madin-Darby canine kidney cells (MDCK1) cells has been previously described in Braun et al. (23). Two days before the experiments, 25,000 MCT8-expressing and 60,000 mock-transfected MDCK1 cells were seeded into 24-well plates. Uptake assays were performed with 10 nM 125I-T3 diluted in DMEM/F12 (1:1) in the presence of 0.1 nM to 400 μM JPH203 for 3 minutes.
Statistics
The results were obtained by combining several independent experiments with samples from different differentiation experiments in one graph. For radioactive uptake measurements, each dot represents the mean of a duplicate normalized to protein content.
The number of repetitions is reported in the figures. ImageJ was used for the quantification of Western blots. We used GraphPad Prism 6 for the generation of graphs and statistical analysis.
Statistical analysis of the 3′-mRNA sequencing data and ribosome profiling data was performed with R and the DESeq2 package for normalization of raw counts and dispersion estimation as recommended by the provider. In addition, we used a negative binomial Wald test with Benjamini–Hochberg multiple test correction for analyses and defined an adjusted q-value <0.05 as significant.
Results
Characterization of iBMECs
We used the following iBMEC lines: iBMECs from a healthy control are termed MCT8 throughout the article. Its isogenic control with an 8 bp-deletion in exon 3 is called MCT8delEx (8). The iBMECs carrying the pathogenic MCT8-P321L mutation are termed P321L. Its isogenic control cell line is called P321Lcorr and was created via the CRISPR/Cas9 system, thereby reverting the mutant leucine codon with wild-type proline (8).
To characterize the barrier formation in differentiated iBMECs, we demonstrated the expression of CLAUDIN-5 as a specific marker for the BBB, OCCLUDIN as a marker for tight junctions, and platelet endothelial cell adhesion molecule-1 as a marker for endothelial cells by immunocytochemical staining (Supplementary Fig. S1A) (28 –30). In addition, we investigated the expression of endothelial genes, which are enriched in iBMECs compared with undifferentiated iPSCs (Supplementary Fig. S1B).
In a previous study (8), transwell experiments have been used to characterize the TH transport across the barrier of iBMECs. The tightness of the barrier was investigated by using fluorescein. Fluorescein is not actively transported across the barrier but can diffuse when the barrier is not tight. Only 4% of the overall fluorescein was detected in the lower compartment for iBMECs expressing functional MCT8 and P321Lcorr, respectively, after 60 minutes of incubation (Supplementary Fig. S1C). Both iBMEC lines were able to transport T3 across the barrier (Supplementary Fig. S1D). We were not able to obtain a tight barrier with iBMECs expressing pathogenic P321L and MCT8delEx (Supplementary Fig. S1E, F).
Investigation of translated mRNAs by ribosome profiling displayed a decreased amount of various ribosome-protected mRNA fragments in P321L-iBMECs coding for proteins that mediate the interaction of cells with components of the extracellular matrix (Supplementary Fig. S1G). These proteins are possibly involved in cell adhesion on the surface of the transwell by binding to fibronectin and collagen IV. Due to the disturbed adhesion found in iBMECs expressing pathogenic MCT8, we performed the following experiments with iBMECs seeded on plates. This allowed us to measure the barrier-independent uptake of TH into iBMECs but not its transport across the barrier.
Reduced TH uptake in iBMECs with pathogenic MCT8 mutations
The iBMECs expressing pathogenic P321L display significantly reduced T3 uptake activities compared with iBMECs expressing healthy control MCT8 (Fig. 1A) (8). Expression studies of MCT8 protein in control- and patient-derived iBMEC compared with their respective isogenic controls (MCT8/MCT8delEx and P321L/P321Lcorr) lead to the following results (Fig. 1B): Healthy control MCT8 expressed in iBMECs had a molecular weight above 55 kDa. The out-of-frame deletion of exon 3 completely abrogates MCT8delEx protein expression. As expected, iBMECs carrying the P321L mutation have a diminished mutant protein expression compared with P321Lcorr.
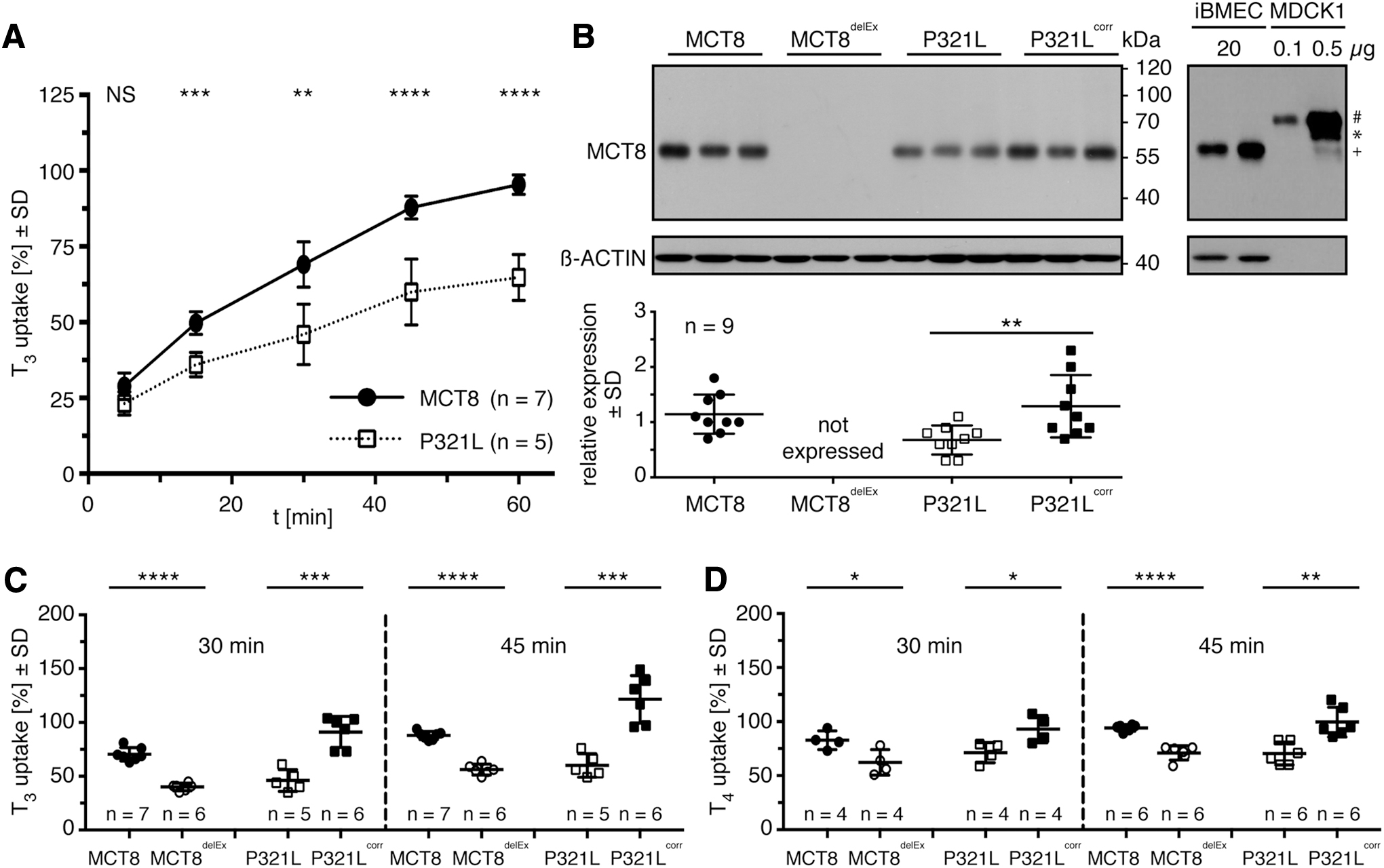
Reduced transport activity of pathogenic MCT8 expressed in iBMECs. (
Further, a comparison of MCT8 expression in iBMECs and MCT8 overexpressing MDCK1 cells demonstrates that human-derived iBMECs express the 56 kDa form of MCT8 (initiating at Met75), while transfected MDCK1 cells predominantly express the long isoform migrating at almost 70 kDa (Fig. 1B).
To investigate the transport activity of pathogenic iBMECs compared with their isogenic controls, we performed T3 and T4 uptake assays (Fig. 1C). P321L-iBMECs showed significantly decreased T3 uptake compared with P321Lcorr-iBMECs. Similar results were obtained for MCT8delEx-iBMECs that do not express MCT8 at all compared with healthy control MCT8-iBMECs.
Transport measurements with T4 as the substrate gave similar results (Fig. 1D): The uptake of T4 is diminished in iBMECs expressing non-functional MCT8 (MCT8delEx-iBMECs, P321L-iBMECs) compared with iBMECs expressing the respective control (MCT8-iBMECs, P321Lcorr-iBMECs).
NaPB rescues T3 uptake in cells expressing pathogenic MCT8
We have previously demonstrated that NaPB rescues the expression of several pathogenic MCT8 mutants when overexpressed in a transfected cell system (19,20). To test whether NaPB can rescue endogenously expressed, pathogenic MCT8 in a physiologically relevant differentiated cell model of human origin, we treated differentiated iBMECs with 2 mM NaPB for 24 hours. Indeed, NaPB treatment of iBMECs expressing P321L and P321Lcorr significantly increased the expression of pathogenic and isogenic control MCT8 protein (Fig. 2A, B).

NaPB rescues expression and T3 transport function of pathogenic and healthy control MCT8. (
Next, we speculated that the increase in protein expression accordingly enhances T3 transport function (Fig. 2C). The left-hand panel shows that SC, a specific MCT8 inhibitor (31), diminishes T3 uptake in P321Lcorr-iBMECs. Moreover, NaPB increases T3 uptake in P321Lcorr cells, and this increase is abrogated by co-incubation with SC. T3 transport into P321L expressing iBMECs is significantly decreased compared with P321Lcorr-iBMECs and not further diminished by co-incubation with SC (Fig. 2C).
T3 uptake of P321L-iBMECs significantly increases after NaPB treatment. This increase is almost entirely abrogated by co-incubation with SC, suggesting that the effect of NaPB on T3 uptake is mediated predominantly by its action on pathogenic P321L (Fig. 2C). However, the incomplete suppression of T3 uptake by SC in NaPB-treated P321Lcorr-iBMECs leaves the possibility for an additional contribution of another RG transporter.
To investigate the possibility of NaPB-dependent induction of another T3 transporter in iBMECs, we compared iBMECs expressing healthy control MCT8 with its isogenic control line MCT8delEx. As seen earlier for P321Lcorr, protein expression of healthy control MCT8 is significantly upregulated in response to NaPB treatment (Fig. 2D, E). This observation indicates that the effect of NaPB on healthy control MCT8 expression is not a clone-specific effect. Similar to P321Lcorr cells, T3 uptake into MCT8-iBMECs significantly decreases on SC incubation to a level found in MCT8delEx-iBMECs (Fig. 2F).
Treatment with NaPB significantly induces T3 transport in healthy control MCT8-iBMECs. The increase in T3 uptake observed for MCT8delEx-iBMECs after NaPB treatment does not reach significance. Similar to the results found for P321Lcorr-iBMECs, the incubation of NaPB-treated MCT8-iBMECs with SC does not reduce T3 transport to the level found for NaPB-untreated MCT8-iBMECs (+ SC) (Fig. 2F). Taken together, the results of the second pair of iBMEC cell lines support the findings that NaPB induces MCT8 expression and transport activity and is capable of inducing another transporter with T3 uptake activity.
Characterization of additional T3 transporters in iBMECs
To gain more information on other TH transporters expressed in the human iBMEC model, we performed 3′-mRNA sequencing and confirmed the expression of SLC16A2 (MCT8), SLC16A10 (MCT10), SLC7A8 (LAT2), and with highest abundance, SLC7A5 (LAT1) while the expression of SLCO1C1 (OATP1C1) was not detectable (Fig. 3A, B) (8). This led us to further investigate the possible involvement of LAT in the background T3 uptake by iBMECs using competition and inhibition studies.

Characterization of additional TH transporters expressed in iBMECs. (
We first focused on LAT1 and LAT2, which are well known for their ability to transport leukine and other neutral amino acids (32). The LAT-mediated transport can be inhibited by BCH (33). Thus, we tested Leu as a competitor and BCH as an inhibitor for LAT-mediated T3 uptake into iBMECs. Both compounds were not able to decrease T3 uptake in all tested iBMEC lines (Fig. 3C, D). JPH203 was reported as a specific inhibitor for LAT1, but it does not significantly inhibit the closely related LAT2 at submicromolar IC50 (34). At 10 μM JPH203 decreased T3 uptake in control iBMECs (Fig. 3C, D).
However, in cells carrying inactivating mutations of MCT8, JPH203 had no significant effect on T3 uptake, suggesting that at the concentration used, it acts as an inhibitor of MCT8. This was confirmed in MCT8-overexpressing MDCK1 cells, where JPH203 inhibits T3 uptake at an IC50 of 12.7 ± 3.9 μM (Supplementary Fig. S2).
We further tested phenylalanine as a competitor that is not only a substrate of the L- but also a substrate of the T-type amino acid transporter a.k.a MCT10 (SLC16A10) (35,36). The reduction of T3 transport in the presence of Phe into iBMECs confirms that Phe competes with T3 for uptake (Fig. 4A, B). Since LATs are not significantly contributing to T3 uptake in iBMEC, we concluded that the inhibition by Phe was likely attributable to MCT10. Unfortunately, we were not able to detect MCT10 by Western blotting to test for an increase in MCT10 expression after NaPB treatment.
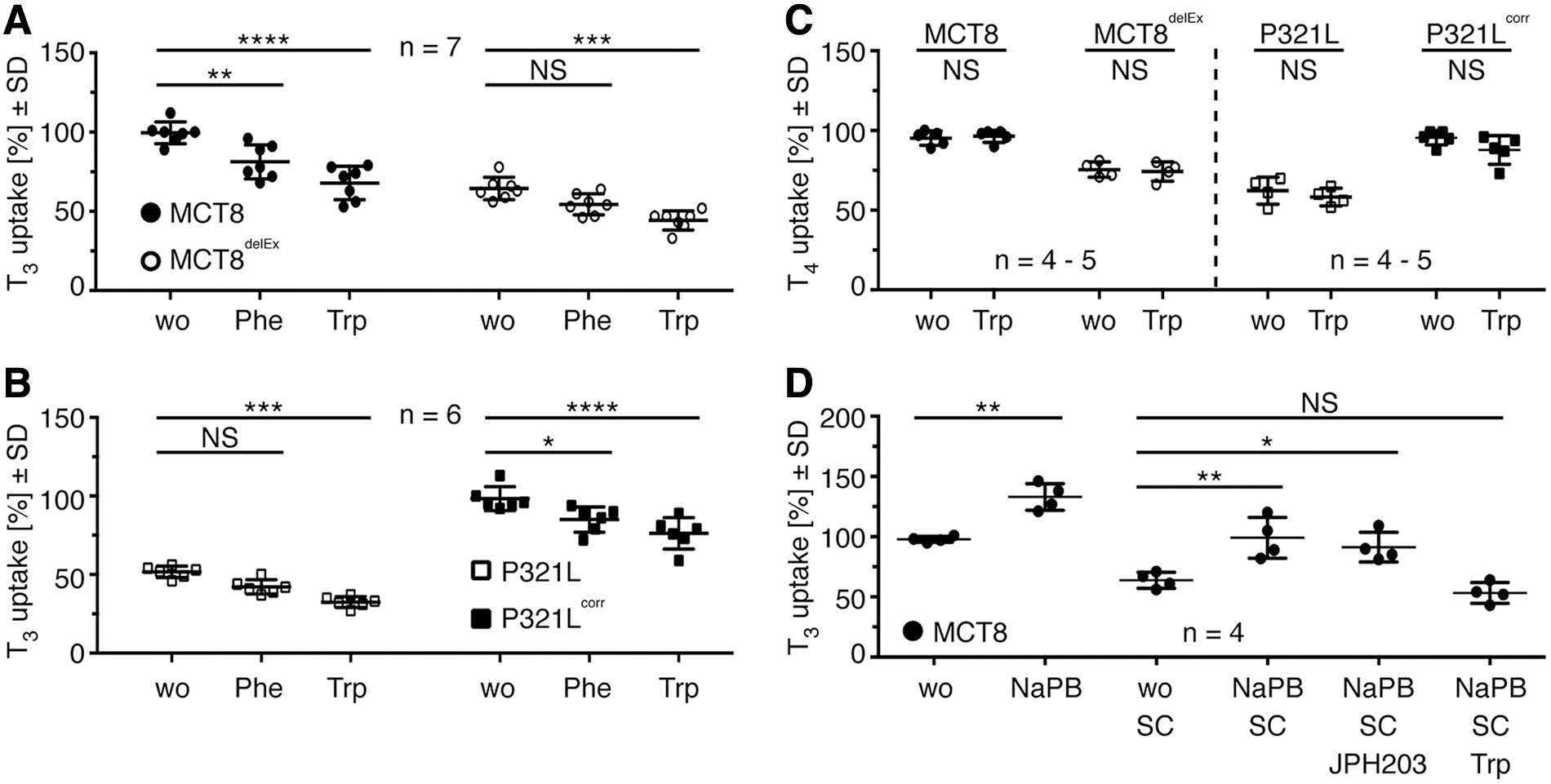
MCT10 is involved in background T3 uptake found in iBMECs. (
We identified an antibody that detected MCT10 in overexpressing insect cells, but the antibody was not sufficient to provide adequate results in iBMECs (Supplementary Fig. S3A). Given that NaPB is described as an inhibitor of class I and class II histone deacetylases (21), we tested for NaPB responsiveness on mRNA levels. We did not detect any differences neither on SLC16A10 nor on SLC16A2 mRNA expression (Supplementary Fig. S3B, C), indicating that class I and class II histone deacetylases are not involved in transcription regulation of MCT10 and MCT8.
MCT10 is also a tryptophan transporter. Hence, we tested whether Trp is able to compete with T3 for uptake into iBMEC lines. Trp competition leads to a significant reduction of T3 uptake in all tested iBMEC lines (Fig. 4A, B). MCT10 is unable to transport T4. Since Trp does not compete with T4 for uptake into iBMECs, we suggest that MCT10 is a second functional T3 transporter in human iBMECs (Fig. 4C). The combination of inhibitors supports the conclusion that MCT10 is an SC-resistant T3 transporter that is also increased by NaPB treatment in iBMEC (Fig. 4D).
NaPB increases T4 uptake in iBMECs
T4 uptake into iBMECs increases significantly after treatment with NaPB (Fig. 5A, B). The increase in T4 uptake observed for NaPB-treated P321L-iBMECs and MCT8delEx-iBMECs even reaches the same levels as in untreated iBMECs expressing functional MCT8 (P321Lcorr-iBMEC and MCT8-iBMEC). Next, we tested whether the increase depends on rescued pathogenic MCT8 expression, the enhanced expression of additional T4 transporters, or both.
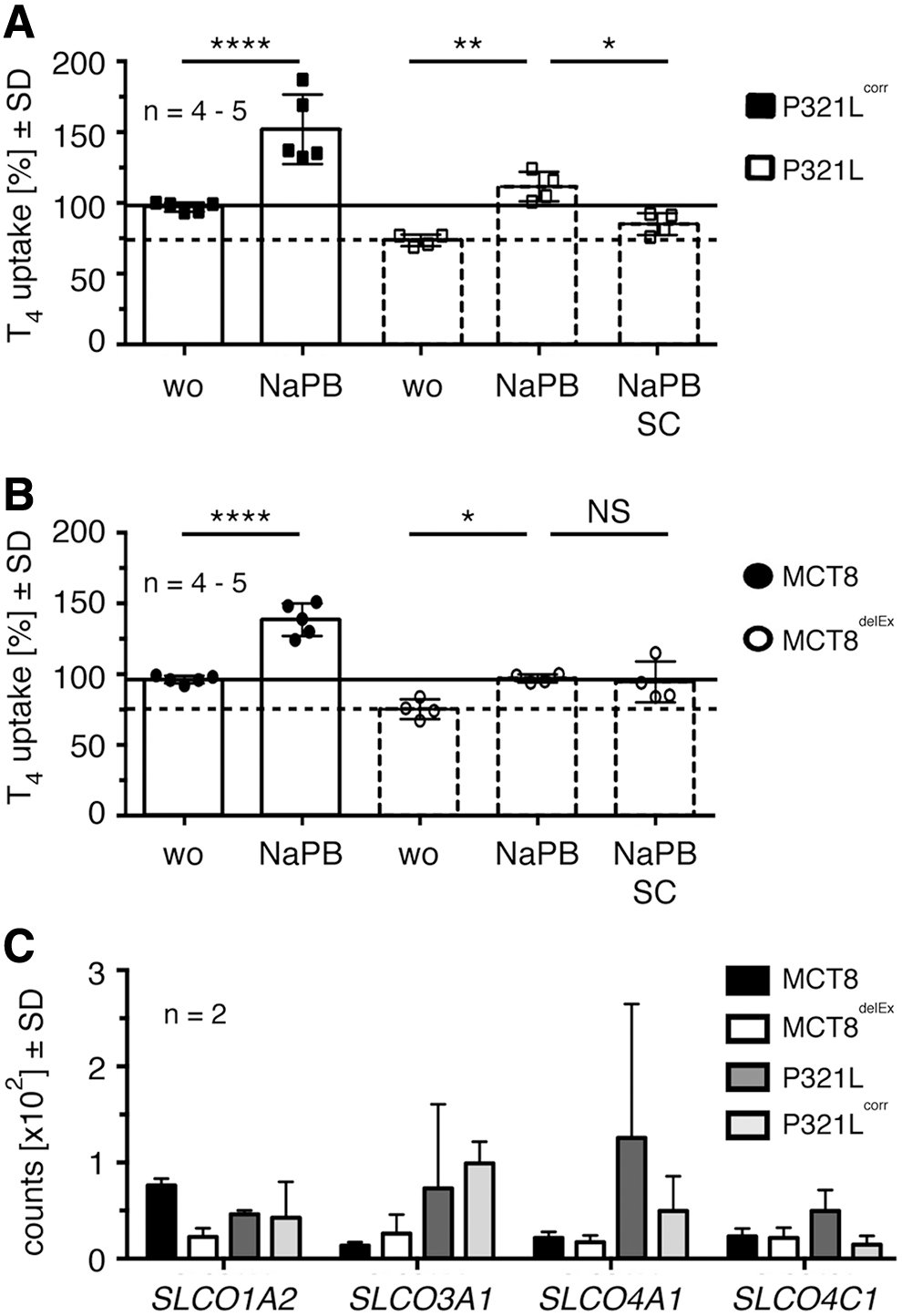
NaPB increases T4 transport function in all tested iBMECs. (
The use of SC significantly decreases T4 uptake in NaPB-treated P321L-iBMECs (Fig. 5A), suggesting that the enhanced T4 uptake after NaPB treatment is partly mediated by rescued pathogenic MCT8. SC does not inhibit the significantly induced T4 uptake in NaPB-treated MCT8delEx-iBMECs (Fig. 5B), pointing toward the involvement of an additional T4 transporter in T4 uptake. 3′-mRNA sequencing data demonstrate the expression of SLCO1A2, SLCO3A1, SLCO4A1, and SLCO4C1 in iBMECs (Fig. 5C). We did not detect the expression of SLCO1B1, SLCO1B3, SLCO1C1 (OATP1C1), SLCO6A1, SLC10A1 (NTCP), and SLC17A4 that are described as T4 transporters (1).
Discussion
We show here that the chemical chaperone NaPB can potentially be used to restore TH transport function by stabilizing pathogenic MCT8 protein expression and activating additional TH transporters in iBMECs.
We recently published that NaPB is able to rescue protein expression of unstable but functional MCT8 mutants in an overexpressing cell system (19,20,24). However, this work included the expression of MCT8 under the control of an artificial promoter in a non-human derived cell model. Here, we addressed both points and chose patient-derived iPSCs differentiated into iBMECs as a model for the BBB (8,37) to test for NaPB responsiveness.
The importance of Mct8 expression at the murine BBB became obvious when Ceballos et al. (10) restored TH content in hypothyroid Mct8-deficient brains with T4 but not T3. T4 import was compensated by Oatp1c1, while T3 uptake exclusively depended on Mct8. Further studies demonstrated the expression of human MCT8 (but not OATP1C1) in enriched BMECs and in microvascular structures of fetal brains (6,7,11,38).
The iBMECs carrying the pathogenic P321L mutation show reduced expression of mutant protein compared with the isogenic control. This observation is contrary to studies in overexpressing cell systems and demonstrates the acute need for working with cells expressing endogenous levels of MCT8. Pathogenic P321L did not show any changes in expression compared with healthy control MCT8 when expressed in COS-1, JEG3, and Flp-in 293 cells (39). We and others hypothesized that the expression and stability of pathogenic MCT8 mutants depend on the cellular context (3,39,40).
In the present work, we used NaPB as a chemical chaperone to rescue decreased pathogenic P321L expression in iBMECs. The use of NaPB as a chemical chaperone was already described for the treatment of cystic fibrosis. Recently, the more specific Food and Drug Administration-approved cystic fibrosis transmembrane conductance regulator (CFTR) modulator Trikafta has been adopted for the treatment of these patients (41). The most frequent mutation in the CFTR protein, ΔPhe508, impairs membrane insertion of the mutant protein and leads to protein degradation (42).
NaPB rescued the mutated CFTR protein and, thus, restored protein function in patients (43,44). Here, we report a similar NaPB-mediated action on pathogenic MCT8 in human iBMECs. NaPB not only rescued expression of the P321L mutant protein, but they also restored T3 and T4 uptake. Similar findings have been made for healthy control MCT8 expressed in iBMECs. In general, proper protein folding is a highly regulated process to maintain proteostasis. It was shown that only 20–25% of wild-type CFTR expressed in COS-7 cells reaches the plasma membrane (45).
The remainder was assumed to remain in the endoplasmic reticulum (ER) and undergo ER-associated degradation due to folding deficits, although not mutated (42). We hypothesize that NaPB assists not only mutated proteins with folding problems but also wild-type proteins, including MCT8 and MCT10, during the complex process of folding.
Inhibition studies with the MCT8-specific inhibitor SC verified that mutant P321L mediates the increase in T3 uptake after NaPB treatment. Further, competition studies with aromatic amino acids suggested the additional involvement of MCT10 in T3 uptake and its upregulation after NaPB treatment, similarly to MCT8. Our findings substantiate the detection of MCT10 in microvessels of human fetal brains (38) as another T3 transporter at the human BBB.
Experiments with T4 support the finding that mutant MCT8 is involved in the rescue of the transport and also suggest an upregulation of additional T4 transporters. We identified several possible candidates. Unfortunately, there are no specific inhibitors available for these transporters to test for their involvement in T4 uptake after NaPB treatment.
Although there are several reports about the beneficial action of NaPB on mouse models with neurodegenerative disorders (46 –48), it is still unclear whether NaPB is able to enter the human brain by crossing the BBB under physiological conditions. Hence, several clinical trials are currently underway to investigate the potential of NaPB in the treatment of these diseases. First, the results on amyotrophic lateral sclerosis display a slower decline in body function and an increase in long-term survival (49,50), which suggest an NaPB-mediated effect on motor neurons in the central nervous system.
In 2016, Lee and Kang (51) showed that Mct1 is responsible for the uptake of NaPB into the rat brain. Whether MCT1 or other MCTs are responsible for the transport of NaPB across the human BBB needs to be elucidated.
The only treatment option for MCT8-deficient patients so far relies on TRIAC that binds to the T3 receptor and is transported across membranes independent of MCT8. TRIAC reversed the hypothyroid brain phenotype in Oatp1c1/Mct8-deficient mice (52). However, it is not reported whether TRIAC treatment cures the locomotor dysfunction reported for these mice (18). TRIAC is currently under investigation in clinical trials ([53] and NCT02396459). Other thyromimetic agents such as sobetirome and its prodrug Sob-AM2 are under preclinical investigation (54).
However, there is the possibility that MCT8 has an additional function beyond T3/T4 transport—either by transporting another substrate or by protein interactions. NaPB, as an approved drug for the chronic use in children (55), might not only restore MCT8-mediated TH transport, but also restore the transport of additional unknown substrates, which may contribute to the symptoms of MCT8 deficiency (e.g., locomotor dysfunction). This cannot be achieved with thyromimetic agents and points to a potential advantage of the NaPB approach.
The NaPB approach only tested one pathogenic MCT8 mutant expressed in iBMECs, and the effect on other MCT8 mutations still needs to be explored. Nevertheless, it may be an example of a proof-of-concept of a potential future personalized medicine for MCT8 deficiency. Further, in the future, patients expressing stable but non-functional MCT8 mutants may also potentially benefit from this treatment due to a possible NaPB-mediated induction of an alternative T3 and T4 transporter at the BBB.
Footnotes
Authors' Contributions
G.D.V. and C.N.S. provided iPS cells. D.B. designed the experiments. D.B. and S.B. performed the experiments. D.B., S.B., and U.S. analyzed the data. D.B. and U.S. wrote the article. G.D.V., C.N.S., and S.B. commented on the article.
Author Disclosure Statement
The authors have declared that no conflict of interest exists.
Funding Information
Financial support was provided by a European Thyroid Association (ETA) research grant, Uniklinikum Bonn and Sherman family foundation.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3