
Abstract
Pathogenic variants in TSHB are known to cause severe isolated central congenital hypothyroidism (CH). In this study, we present the clinical, biochemical, and genetic features of the first patient with a mild central CH phenotype. We identified a novel homozygous variant in TSHB: (Chr1: NM_000549.5):c.290A>G p.(Tyr97Cys) in a newborn girl detected by neonatal CH screening, whose central CH was initially overlooked because of misinterpretation of her plasma-free thyroxine (fT4) concentration. This report adds to the phenotypic spectrum of TSHB variants and underlines the importance of using age-specific fT4 reference intervals to diagnose central CH.
Introduction
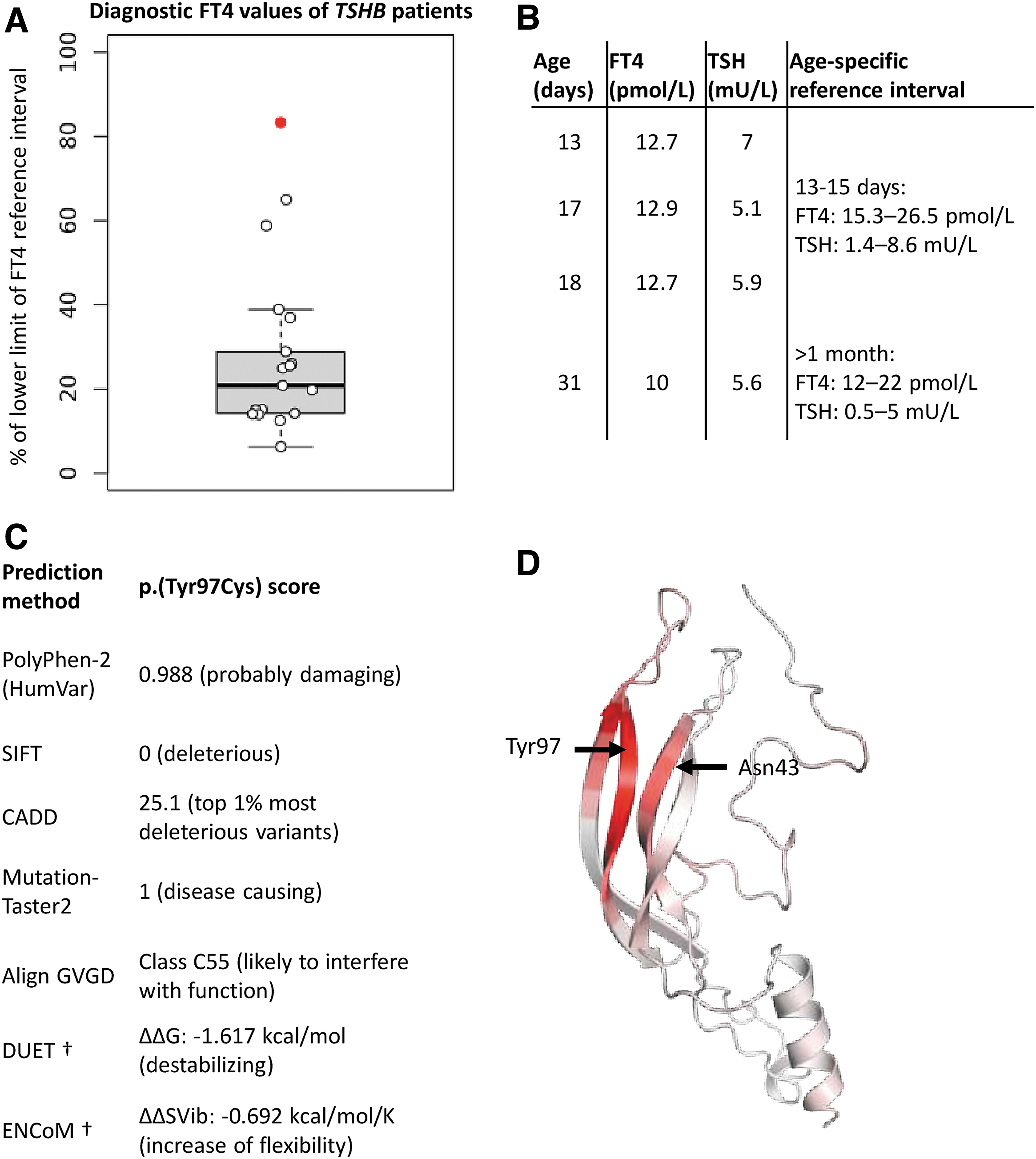
The first gene associated with isolated central congenital hypothyroidism (CH) was TSHB (OMIM #188540), which encodes the beta-subunit of thyrotropin (TSHβ). Since its discovery, about 30 patients with biallelic pathogenic variants have been described. The phenotype is characterized by a severely decreased plasma-free thyroxine (fT4), and an undetectable to mildly elevated TSH level. Until now, the highest recorded value of fT4 at diagnosis was 65% of the lower limit of the reference interval (RI), whereas most patients present with an fT4 around 25% of the lower limit of the RI (Fig. 1A) (1). In this report, we describe a newborn girl with a novel homozygous pathogenic variant in TSHB, manifesting as mild isolated central CH.
(
Case Presentation
The proband is the second-born daughter to consanguineous Pakistani parents (first cousins). She was born at 38 weeks of gestation after a pregnancy complicated by umbilical cord compression. After a first inconclusive result (thyroxine [T4]/thyroxine binding globulin [TBG] ratio below 17), the second neonatal CH screening on the 10th day of life was abnormal. The total T4/TBG ratio (an indirect measure of fT4) was again decreased, while TSH was 2 mIU/L (normal) on both occasions (Supplementary Table S1).
Also, in the first days of life the mother reported sleepiness and poor drinking of her daughter. The girl was referred to a pediatric clinic for further evaluation. During admission sleeping and drinking improved, and she was discharged after two days. Thyroid function tests (Fig. 1B) were erroneously interpreted as normal because of incorrect comparison of neonatal fT4 to the adult RI. In fact, based on the age-specific RI (2), they were highly suggestive of central CH in the absence of illness (nonthyroidal illness syndrome), maternal thyroid disease, and prematurity (transient hypothyroxinemia of prematurity).
At age one month the girl was again admitted because of poor drinking. This time fT4 was (even) below the adult RI (Fig. 1B). IGF-1 and prolactin levels were normal, and cortisol was maximally 750 nmol/L in a high dose adrenocorticotropic hormone test, raising suspicion of isolated central CH. Levothyroxine (LT4) treatment and next-generation sequencing with targeted gene panel analysis (including TSHB, TRHR, TRH, IGSF1, TBL1X, and IRS4) were initiated. DNA was extracted from a peripheral blood sample. We identified a novel homozygous variant in TSHB: (Chr1:NM_000549.3):c.290A>G p.(Tyr97Cys) (for electropherograms see Supplementary Fig. S1).
The proband's parents were heterozygous carriers of the TSHB variant, while her older sister did not carry a pathogenic variant. Thyroid function tests were normal in the proband's family members (Supplementary Table S2).
While treatment with LT4 (started at age 33 days) normalized biochemical indices of thyroid function, unfortunately it did not influence the poor drinking. Although the girl had no other clinical abnormalities (normal growth and development), she refused to drink for which feeding by nasogastric tube was started. After the introduction of solid foods from age six months it was possible to gradually wean her of tube feeding. Now, at the age of 10 months, removal of the tube is pending.
In silico analysis
p.(Tyr97Cys) is detected once in gnomAD v2.1.1 in a heterozygous state in a South Asian man (dbSNP rs777736814). Pathogenicity scores, including protein stability and flexibility analyses, indicated deleteriousness of the variant (Fig. 1C). To gain more insights into the pathogenicity of the variant, we performed in silico analyses with a computed Phyre2 model of TSHβ. There were no steric clashes or structural deficits upon mutation. p.(Tyr97Cys) did not influence the normal disulfide bonds between cysteine residues, which control protein folding of TSHβ and heterodimerization with TSHα. However, the variant increased flexibility of the Tyr97-surrounding area (Fig. 1D). Notably, this region contains Asn43, which is involved in N-acetylglucosamine binding (NAG; implicated in TSHβ glycosylation).
Discussion
Biallelic pathogenic variants in TSHB are known to cause severe isolated CH (1). In this study, we present the medical history of a patient with a mild phenotype (Fig. 1A) and the homozygous variant p.(Tyr97Cys). We speculate this discrepancy may be explained by differences between previously reported variants and p.(Tyr97Cys). Known variants either lead to a complete loss of TSHβ (whole gene deletion, premature termination codon and start loss variants) or structural deficits that completely impair TSH function; for example, p.(Gly49Arg), which destabilizes a critical area for dimerization with TSHα, or p.(Cys105Arg) and p.(Cys108Tyr), which lead to loss of disulfide bond-forming cysteine residues.
Based on the clinical case of the proband, the p.(Tyr97Cys) variant presumably leads to a combined mild quantitative and mild qualitative deficiency of TSH. This may be inferred from the relationship between TSH and fT4 in the proband. TSH was in the normal range, while fT4 was decreased, which supports the hypothesis of a quantitative deficiency. Furthermore, the fact that TSH was (well) within normal ranges—at the first investigation even near the upper limit of the RI—also supports the hypothesis of a qualitative TSH problem.
We hypothesize that the quantitative deficiency arises due to increased denaturation, as a consequence of decreased protein stability. The qualitative deficiency possibly results from heterodimerization defects with TSHα, as hypothesized in other TSHB-related reports (3), due to abnormal protein folding of TSHβ. Also, p.(Tyr97Cys) regionally alters protein flexibility and affects Asn43 (Fig. 1D), possibly influencing its capacity to bind NAG. It was reported that experimental removal of NAG from TSH decreased its bioactivity significantly (4). In view of these observations, we conclude that the mild central CH phenotype is caused by the homozygous p.(Tyr97Cys) TSHB variant.
In conclusion, we report a novel homozygous TSHB variant p.(Tyr97Cys) in an individual with central CH. This report adds to the phenotypic spectrum of TSHB variants, since it describes the first patient with a mild phenotype. Furthermore, this case underlines the importance of using age-specific RIs of fT4 to diagnose central CH.
Footnotes
Acknowledgment
We are grateful to the family for participating in this study.
Authors' Contributions
The first draft of the article was written by P.L., and all authors commented on subsequent versions of the article. A.B. authorized the biochemical analyses. H.B. performed genomic analyses. P.L. performed in silico analyses. J.J.J. and N.Z.-S. were involved in patient care. The project was supervised by A.S.P.v.T. and N.Z.-S. All authors read and approved the final article and confirm authorship.
Statement of Ethics
The proband and her family agreed to participate and signed informed consent forms for publication of clinical and genetic data. This study was approved by the medical ethical committee of Amsterdam UMC, location AMC (METC AMC), project number W21_393 # 21.438.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2