
Abstract
Background:
Medullary thyroid cancer (MTC) is a rare malignancy originating from the calcitonin-producing C cells of the thyroid. Despite recent therapeutic advances, metastatic MTC remains incurable. Adoptive cell therapy (ACT) using genetically engineered T cells targeting either tissue-restricted tumor-associated antigens or mutated neoantigens has led to durable remissions in other metastatic solid tumors. The majority of MTC express the tumor-associated antigens calcitonin and carcinoembryonic antigen (CEA), and ∼40% of MTC harbor the RET M918T oncogenic driver mutation.
Methods:
We developed and characterized three immunoreceptors that recognize extracellular CEA, a calcitonin epitope presented by HLA-A*24:02, or an RET M918T neoepitope restricted by HLA-DPB1*04:01/02. The chimeric antigen receptor (CAR) targeting CEA was synthetically designed, while the T cell receptors (TCRs) targeting calcitonin and RET M918T were isolated from a transgenic mouse and patient with MTC, respectively. These immunoreceptors were genetically engineered into peripheral blood T cells and tested for antigen specificity and antitumor activity.
Results:
T cells expressing the anti-CEA CAR or the calcitonin-reactive TCR produced effector cytokines and displayed cytotoxicity against cell lines expressing their cognate antigen in vitro. In immunodeficient mice harboring a human MTC cell line, the adoptive transfer of T cells engineered to express the anti-CEA CAR or calcitonin-reactive TCR led to complete tumor regression. T cells expressing the HLA-DPB1*04:01/02-restricted TCR targeting RET M918T, which was cloned from peripheral blood CD4+ T cells of a patient with MTC, demonstrated specific reactivity against cells pulsed with the mutated peptide and MTC tumor cells that expressed HLA-DPB1*04:01 and RET M918T.
Conclusion:
The preclinical data presented herein demonstrate the potential of using genetically engineered T cells targeting CEA, calcitonin, and/or RET M918T to treat metastatic MTC.
Introduction
Medullary thyroid cancer (MTC) is a rare neuroendocrine tumor, which originates from the calcitonin-secreting parafollicular C cells of the thyroid. MTC comprises only 2–5% of all thyroid cancers but accounts for a disproportionate number of thyroid cancer deaths. 1,2 Approximately 40–50% of MTC tumors have mutations in the RET oncogene, which is associated with a less favorable prognosis compared with wild-type (WT) RET tumors. 3 Once MTC is metastatic, radiation therapy and RET kinase inhibitors can prolong patient survival but are not curative. 4 –8 Complete surgical resection is currently the only curative therapy, as MTC cells do not uptake radioactive iodine. 9
Great advances have been made toward leveraging the immune system to eradicate solid tumors. Immune checkpoint inhibitors (ICI) targeting the PD-1 and CTLA-4 pathways have produced durable complete responses and potential cures in many patients. 10,11 Evidence suggests that at least some of these clinical responses were due to the activation of T cells that targeted mutated antigens (neoantigens) expressed by the patient's tumor. 12,13 Indeed, ICI therapy has the greatest efficacy in solid cancers which tend to have high tumor mutation burdens, 14,15 as only ∼1% of somatic mutations are thought to be immunogenic. 16 The very low mutation burden in medullary thyroid tumors (17.9 somatic mutations on average 17 ) restricts the number of potential neoantigens that can be recognized by endogenous T cells, which is predicted to limit the efficacy of ICI therapy in patients with MTC. Therefore, novel approaches are likely required to more effectively harness T cell-based immunity against MTC. 18
In adoptive cell therapy (ACT), patients can be infused with ∼10 6 –10 11 tumor-reactive T cells, as a means to overcome a weak or absent endogenous antitumor T cell response. The transfer of ex vivo-expanded tumor-infiltrating lymphocytes (TIL) or T cells genetically engineered to express antitumor chimeric antigen receptors (CAR) or T cell receptors (TCRs) has mediated durable cancer regression in some patients with solid tumors 19,20 but has not been evaluated in patients with MTC. The selection of tumor antigen to target is a critical parameter in ACT. When present, antigens derived from mutations, known as neoantigens, represent ideal and safe targets since they are truly tumor-specific.
However, targeting neoantigens may be challenging in MTC given their typically low mutation burden. Other antigens that have been targeted with ACT include tumor-associated self-antigens (e.g., CD19, BCMA, carcinoembryonic antigen [CEA], GD2, AFP, and HER2) and cancer-germline antigens (e.g., NY-ESO-1, and MAGE-A3). 19 –25 In particular, anti-CD19 and anti-BCMA CAR-T cells have revolutionized the standard of care for B cell malignancies and multiple myeloma, highlighting the therapeutic potential of targeting tissue-restricted tumor antigens in addition to neoantigens. 26,27 However, caution is warranted when targeting self-antigens due to the strong potential for on-target/off-tumor toxicities. 24,28 –30
A recent immune profiling study of MTC demonstrated low and variable expression of cancer-germline antigens but found that the tumor-associated antigens CEA (CEACAM5) and calcitonin were highly expressed in nearly all tumor specimens, with CEA exhibiting 3+ staining in all but one primary tumor and calcitonin (CALCA) messenger RNA (mRNA) levels exceeding the housekeeping gene beta actin (ACTB) in most patients. 31 Notably, CALCA and CEA mRNA levels were substantially higher than GFRA4, another MTC-expressed self-antigen that has been targeted in preclinical models using anti-GFRA4 CAR-T cells. 32 Indeed, calcitonin and CEA are well-known serum tumor markers for MTC, which tend to increase with tumor burden, 33 and the application of anti-CEA CAR-T cells to treat MTC has been previously postulated. 18
As a step toward advancing ACT in MTC, we developed both self-antigen and neoantigen-specific immunoreceptors. Herein, we describe the generation and characterization of three immunoreceptors, each targeting a unique MTC antigen. The first receptor, Cal24TCR, is an HLA-A*24:02-restricted TCR that specifically recognizes a 9 amino acid long (9mer) epitope of calcitonin. The second receptor, MT-CAR, is a CAR targeting CEA. The third receptor, RET-TCR, is an HLA-DPB1*04:01/02-restriced TCR targeting the common RET M918T driver mutation, which is found in 30–40% of sporadic MTC. 3 These immunoreceptors expand the range of addressable MTC antigens and provide additional avenues for the development of novel T cell-based therapies to treat MTC.
Materials and Methods
A detailed and comprehensive materials and methods can be found in the Supplementary Methods S1. A summary of the immunoreceptors and experiments performed in this study can be found in Table 1. A brief overview of methods is provided as follows.
Summary of the Immunoreceptors Used and Experiments Performed in This Study
Transgenic HLA-A*24:02 mouse.
Patient with RET-M918T-positive MTC.
ACT, adoptive cell therapy; CAR, chimeric antigen receptor; CEA, carcinoembryonic antigen; MTC, medullary thyroid cancer; TCR, T cell receptor.
Cell lines
The TT and MZ-CRC-1 (MZ) MTC cell lines were provided by Dr. Jena French (UC-Denver), and all other cell lines were purchased from ATCC. In some instances, cell lines were transduced to co-express HLA-A*24:02 using a custom lentiviral vector. We term the HLA-A24 expressing MTC cells lines as TT-A24 and MZ-A24.
Generation of the MHC class I HLA-A24 restricted calcitonin-specific TCR, Cal24TCR
HLA-A24 transgenic mice (Taconic) were immunized with calcitonin protein. Following sacrifice, splenocytes were isolated and then in vitro stimulated with the calcitonin peptide TYTQDFNKF (LifeTein). Following stimulation, calcitonin-reactive T cells were isolated by fluorescence-activated cell sorting (FACS) and underwent TCR sequencing (iRepertoire) to identify the dominant alpha and beta chains.
Generation of the anti-CEA CAR MT-CAR
The sequence of the anti-CEA CAR was generously provided by Dr. Owen Witte (UCLA). Several genetic modifications to the intracellular signaling domain were made to this CAR as described in the Results section.
Generation of the MHC class II restricted RET M918T-specific TCR, RET-TCR
A patient with RET M918T-positive MTC who was successfully being treated with the RET inhibitor pralsetinib was enrolled onto the Portland Medical Center Institutional Review Board approved protocol number 06-108A. To isolate RET M918T-specific TCRs, we in vitro stimulated peripheral blood mononuclear cells (PBMCs) from this patient for 14 days and then evaluated reactivity via IFN-γ enzyme-linked immunosorbent spot assay (ELISPOT) by coculturing with RET M918T peptide-pulsed or RET M918T mRNA-pulsed antigen-presenting cells (dendritic cells, B cells, or PBMCs). WT RET peptides or mRNA were used as controls. CD4+ T cells recognizing RET M918T were subsequently sorted by FACS and their TCRs were sequenced.
Transduction of T cells with MT-CAR, Cal24TCR, and RET-TCR
We utilized the MSGV1 vector system and 293GP cells to generate retroviral supernatants that were used to transduce human T cells. PBMC were stimulated with OKT3 (50 ng/mL) or CD2/CD3/CD28 Activator (StemCell Technologies) and then transduced 48 hours later on retroviral-coated plates, followed by expansion with IL-2 (300 IU/mL) in 50/50 media for 10–14 days. Transduction efficiency was validated by flow cytometry staining. Cal24TCR cells were further expanded with irradiated PBMCs (feeders), IL-2 (3000 IU/mL), and OKT3 (30 ng/mL) using a rapid expansion protocol (REP).
Characterization of MT-CAR, Cal24TCR, and RET-TCR
Flow cytometry was used to evaluate differentiation state, activation state, and intracellular cytokine production of MT-CAR-, Cal24TCR-, and RET-TCR-transduced T cells. Additional functional activity and avidity were measured using IFN-γ ELISPOT analysis. Direct cytotoxic capability of MT-CAR- and Cal24TCR-transduced T cells against tumor cells in vitro was quantified using the Incucyte live-cell imaging system. The MHC class II restriction element for RET-TCR was confirmed by transfecting allogeneic B cells with the donor's MHC class II alleles and evaluating for reactivity via flow cytometric analysis of activation markers and IFN-γ ELISPOT.
Xenogeneic mouse model
NSG mice were inoculated in the hind flank with TT-A24 cells at Rincon Bio under IACUC-approved protocols. After tumors reached ∼125 mm3, mice were randomized into three groups and treated twice at weekly intervals with 5 million Cal24TCR T cells, MT-CAR T cells, or nontransduced T cells (control arm) followed by three daily injections of i.p. 100,000 IL-2 support. Tumors were measured twice weekly and mice were weighed weekly and observed for signs of distress. Tumor curves were compared using the Wilcoxon rank sum test. Experiments were performed in accordance with Arrive 2.0 guidelines.
Impact of the RET inhibitor pralsetinib on Cal24TCR and MT-CAR MTC cell line recognition and impact of pralsetinib on in vitro cell viability
TT-A24 cells were pretreated for 72 hours with varying concentrations of pralsetinib and then cocultured with T cells. After 24 hours, T cell reactivity was characterized by staining T cells for the activation marker CD137.
Results
Generation and characterization of an HLA-A*24:02-restricted TCR targeting calcitonin
The Immune Epitope Database (IEDB;
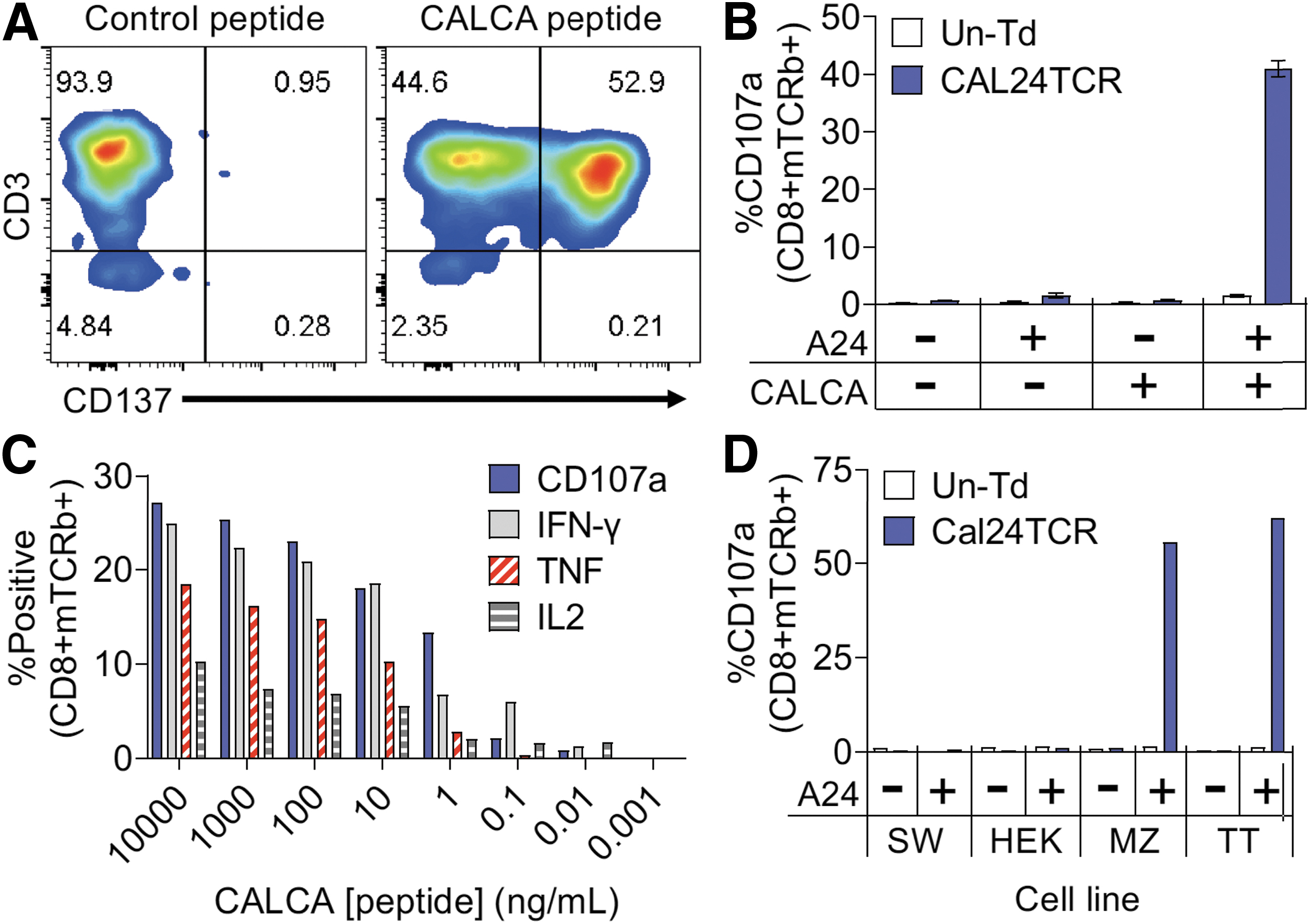
Development and characterization of calcitonin-specific TCR, Cal24TCR. (
To validate the specificity and HLA restriction of Cal24TCR cells, HEK293T cells were transfected with plasmids encoding HLA-A24 alone, the entire CALCA protein alone, or both proteins followed by coculture with the Cal24TCR-transduced T cells. As shown in Figure 1B, T cell reactivity was observed in the TCR-transduced T cells only when HEK293T cells were transfected with both the HLA-A24 (A24) and CALCA genes. We did not observe CD107a or CD137 expression in untransduced T cells or the TCR-transduced CD4+ population (data not shown), which implies that the Cal24TCR requires CD8 co-receptor binding to function.
We next tested the functional avidity of the Cal24TCR by coculturing Cal24TCR T cells from an HLA-A24 + donor with HLA-A24-transfected HEK293T cells pulsed with varying concentrations (10 μg/mL to 1 pg/mL) of TYTQDFNKF peptide. After 6 hours of coculture, cells were evaluated for expression of the degranulation marker CD107a and effector cytokines IFN-γ, IL-2, and TNF by intracellular cytokine staining. As shown in Figure 1C, Cal24TCR could recognize peptide at concentrations down to about 10 pg/mL with an EC50 value of 1–10 ng/mL, which is an avidity level similar to other TCRs that have demonstrated clinical activity. 34
To determine whether Cal24TCR could recognize MTC tumor cell lines, which express calcitonin, we cocultured Cal24TCR T cells with TT and MZ MTC cells as well as the non-MTC cell lines HEK293T and SW480 that were either WT or transduced to express HLA-A24. Figure 1D demonstrates that the Cal24TCR T cells degranulated only when cocultured with the MTC cell lines that expressed HLA-A24. Notably, there was no reactivity to WT HEK293T or SW480 cells or HLA-A24-transduced HEK293T or SW480 cells. Collectively, this data set indicates that Cal24TCR recognizes the minimal TYTQDFNKF calcitonin epitope at concentrations down to 10 pg/mL and that this epitope is naturally processed and presented on HLA-A24 by calcitonin-producing MTC cells.
Development and characterization of MT-CAR targeting CEA
The previously described anti-CEA CAR employing a labetuzumab scFv, 35 long IgG4 spacer, and CD28 costimulatory domain was selected for further development, as the labetuzumab scFv has previously been employed in clinical studies in both antibody-drug conjugate and radioligand formats. 36,37 We made several modifications to this CAR. First, L235E and N297Q mutations were incorporated into the IgG4 spacer to reduce FcγR binding, which can lead to harmful nonspecific CAR activation via cross-linking from pulmonary macrophages and other FcγR expressing cells. 38 Second, we substituted the 28z costimulatory domain with the Stat3/Stat5 activating 28-ΔIL2RB-z (YRHQ) costimulatory domain, which has conferred superior performance in preclinical tumor models compared with the 28z domain. 39 We term the labetuzumab scFv-based CAR with these modifications for applications in MTC as MT-CAR (Fig. 2A), and this CAR was transduced to high efficiencies in peripheral blood T cells (Supplementary Fig. S1).

Development and characterization of anti-CEA CAR MT-CAR. (
To test the specificity of MT-CAR, we cocultured T cells transduced to express MT-CAR with the CEA-negative MiaPaca2 pancreatic cancer cell line that was transfected to express CALCA or CEACAM5 (CEA), or not transfected (Lipofectamine only control). As shown in Figure 2B, MT-CAR-transduced T cells only upregulated CD137 in response to MiaPaca2 cells that had been transfected with CEACAM5. Next, we cocultured MT-CAR cells with the CEA-positive cell lines BxPC3, MZ, and TT and the CEA-negative cell lines SW480 and MiaPaca2 (Supplementary Methods and Supplementary Fig. S2A). Robust CD137 expression (30–80%) was observed on CD8 and CD4 MT-CAR T cells in response to the CEA-positive TT, MZ, and BxPC3 cells lines, while only a minimal response (2–3%) was observed against the CEA-negative SW480 and MiaPaca2 cell lines (Fig. 2C).
Characterization and comparison of Cal24TCR and MT-CAR T cells
Cal24TCR and MT-CAR cells were cocultured with the antigen-positive MTC cells lines (MZ-A24 and TT-A24) or antigen-negative MiaPaca2 cells for 6 hours, followed by evaluation of CD107a, IFNγ, IL-2, IL-4, and TNF expression using flow cytometry. As shown in Figure 3A–C, MT-CAR and Cal24TCR cells produced multiple effector cytokines in response to the antigen-positive MTC cell lines, whereas <2% of the cells produced cytokines when cultured with the antigen-negative MiaPaca2-A24 (MP-24) cell line. As expected, a higher percentage of MT-CAR cells stained positive for IFNγ, TNF, and IL-2 when cocultured with TT-A24 cells than MZ-A24 cells owing to the higher expression of CEA (antigen density) present on TT-A24 cells (Supplementary Fig. S2A). The cytokine response of Cal24TCR CD8 cells was similar for both MTC cell lines, which may be due to both cell lines producing significant levels of calcitonin.
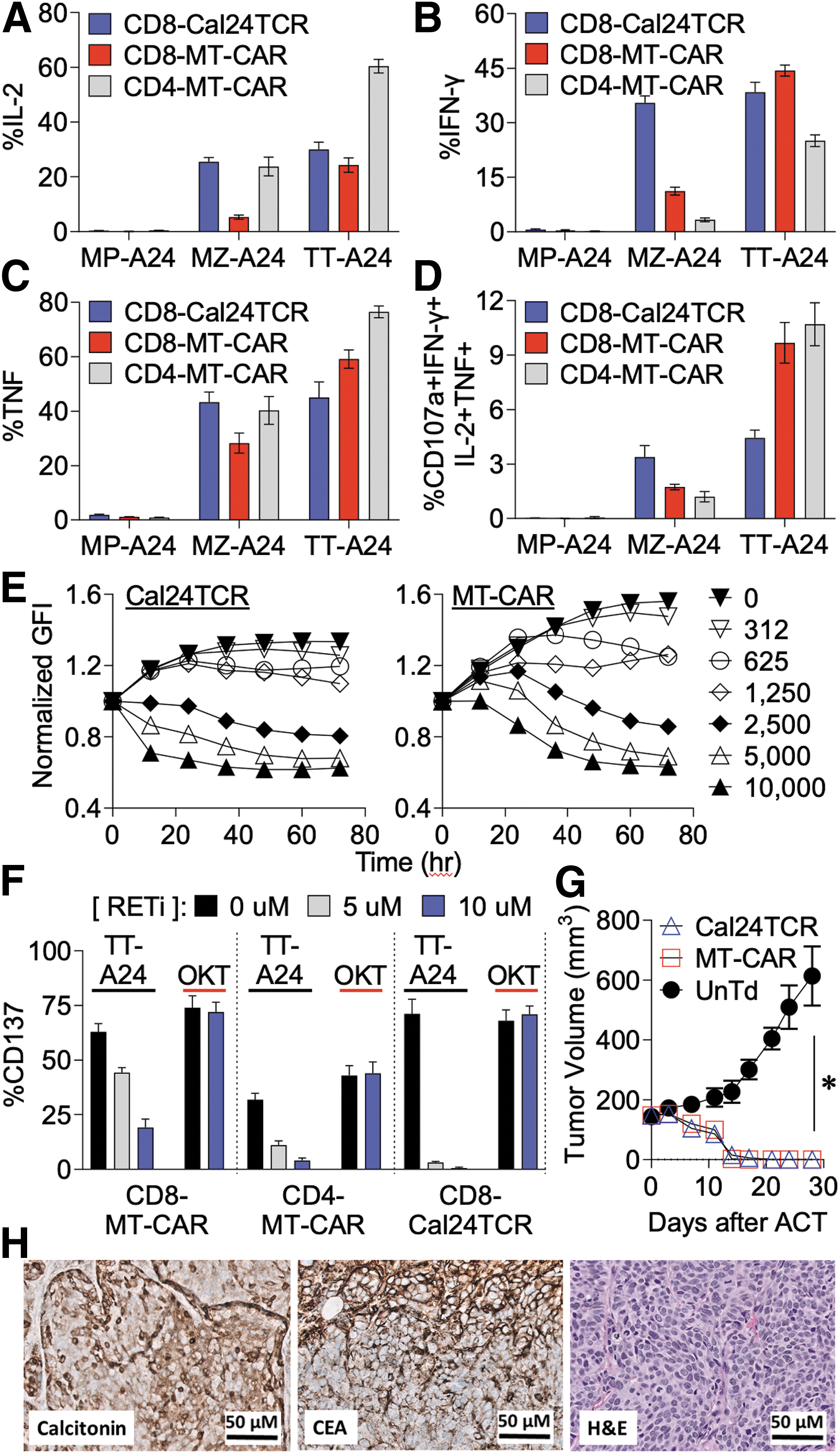
Cal24TCR and MT-CAR recognize and kill MTC cells in vitro and in vivo. (
Little IL-4, which can be an immunosuppressive cytokine, was produced by MT-CAR and Cal24TCR (Supplementary Fig. S3). We found that ∼2–4% of CD8 Cal24TCR and 9–11% of MT-CAR cells were CD107a+IL-2+IFNγ+TNF+ after stimulation with TT-A24 cells (Fig. 3D). MT-CAR cells exhibited increased expression (median fluorescence intensity [MFI]) of all effector cytokines in response to TT-A24 cells relative to MZ-A24 cells, whereas the MFI response magnitude of Cal24TCR CD8 T cells was nearly equivalent for both cell lines (Supplementary Fig. S4). Of potential translational relevance, we found that incubation of MTC cell lines with the RET inhibitor Gavreto (pralsetinib) abolished recognition by Cal24TCR T cells and diminished recognition by MT-CAR (Fig. 3F) likely due to the known effect of pralsetinib on decreasing CEA and calcitonin levels, 40,41 while pralsetinib did not impact TCR signaling mediated by anti-CD3 antibody (Supplementary Fig. S5).
We next used the Incucyte live cell imaging system to monitor killing of either 10,000 TT-A24 or MZ-A24 MTC cells by MT-CAR and Cal24TCR cells over a period of 3 days and generated time-lapse videos of cell killing. As shown in Figure 3E, both Cal24TCR (left) and MT-CAR (right) T cells could eradicate 10,000 TT-A24-eGFP cells as indicated by the reduction of green fluorescence protein intensity when cultured at effector:target (E:T) ratios ≥1:4, corresponding to 2500 T cells per well. Viable TT-A24 cells were observed at 72 hours for wells seeded with <2500 T cells. To eradicate the MZ-A24 cell line, a higher E:T ratio (1:2) was required for MT-CAR cells (5000 total cells) than Cal24TCR cells, which only required a 1:16 E:T ratio (625 total cells).
Notably, untransduced T cells did not kill tumor cells (Supplementary Fig. S6), demonstrating that the response was not mediated by alloreactivity and required MT-CAR or Cal24TCR expression. Neither MT-CAR nor Cal24TCR could control the growth of the antigen-negative MiaPaca2 cell line in vitro (Supplementary Fig. S7). Time-lapse videos of both 625 Cal24TCR cells and 2500 MT-CAR cells killing MZ-A24 cells are provided for direct visualization of the dynamic tumor cell killing process (see links in Supplementary Figs. S6 and S12). Interestingly, high concentrations of the RET inhibitor pralsetinib appeared to mediate cytostatic, rather than cytotoxic, effects on the TT cell line in vitro (Supplementary Fig. S8).
Cal24TCR and MT-CAR T cells eradicate MTC tumors in mice
After establishing in vitro proof of concept, we conducted a murine xenogeneic in vivo study to further assess the preclinical activity of MT-CAR and Cal24TCR. NSG mice were inoculated in the hind flank with the TT-A24 cell line, and 18 days later were adoptively transferred with 5 million untransduced, Cal24TCR, or MT-CAR T cells on treatment days 0 and 7 along with IL-2 support on treatment days 0, 1, 2, 7, 8, and 9 (Supplementary Methods S1).
As shown in Figure 3G, both MT-CAR and Cal24TCR T cells demonstrated potent efficacy, inducing complete tumor regressions in 100% of treated mice (3/3 in each group), whereas tumors uniformly progressed in mice treated with untransduced control cells during the 4-week study period. No significant difference in efficacy was noted between MT-CAR and Cal24TCR cells. The implanted tumors were confirmed to uniformly express the targeted antigens CEA and calcitonin as determined by immunohistochemistry (Fig. 3H). No observable toxicity or weight loss was observed in mice (Supplementary Fig. S9), but on-target/off-tumor toxicity could not be evaluated since normal tissues from NSG mice do not express the targeted antigens.
Isolation, cloning, and validation of the RET M918T-reactive TCR, RET-TCR
Mutated neoantigens derived from hotspot mutations represent ideal antigens to target due to their biological importance and tumor-specific expression. Thus, we tested whether we could identify TCRs that target the RET M918T hotspot mutation, which is frequently expressed in MTC. To that end, PBMCs from an RET M918T+ sporadic MTC patient underwent an in vitro stimulation process using RET M918T long peptide and compounds that activate innate immunity. 42 After 2 weeks, we tested the T cells for reactivity against RET M918T. We detected CD4+ T cells that upregulated 4-1BB (CD137) upon stimulation with the RET M918T RNA transfected autologous antigen presenting cells (APCs) (Fig. 4A), while no reactivity was detected in the CD8+ population (data not shown).
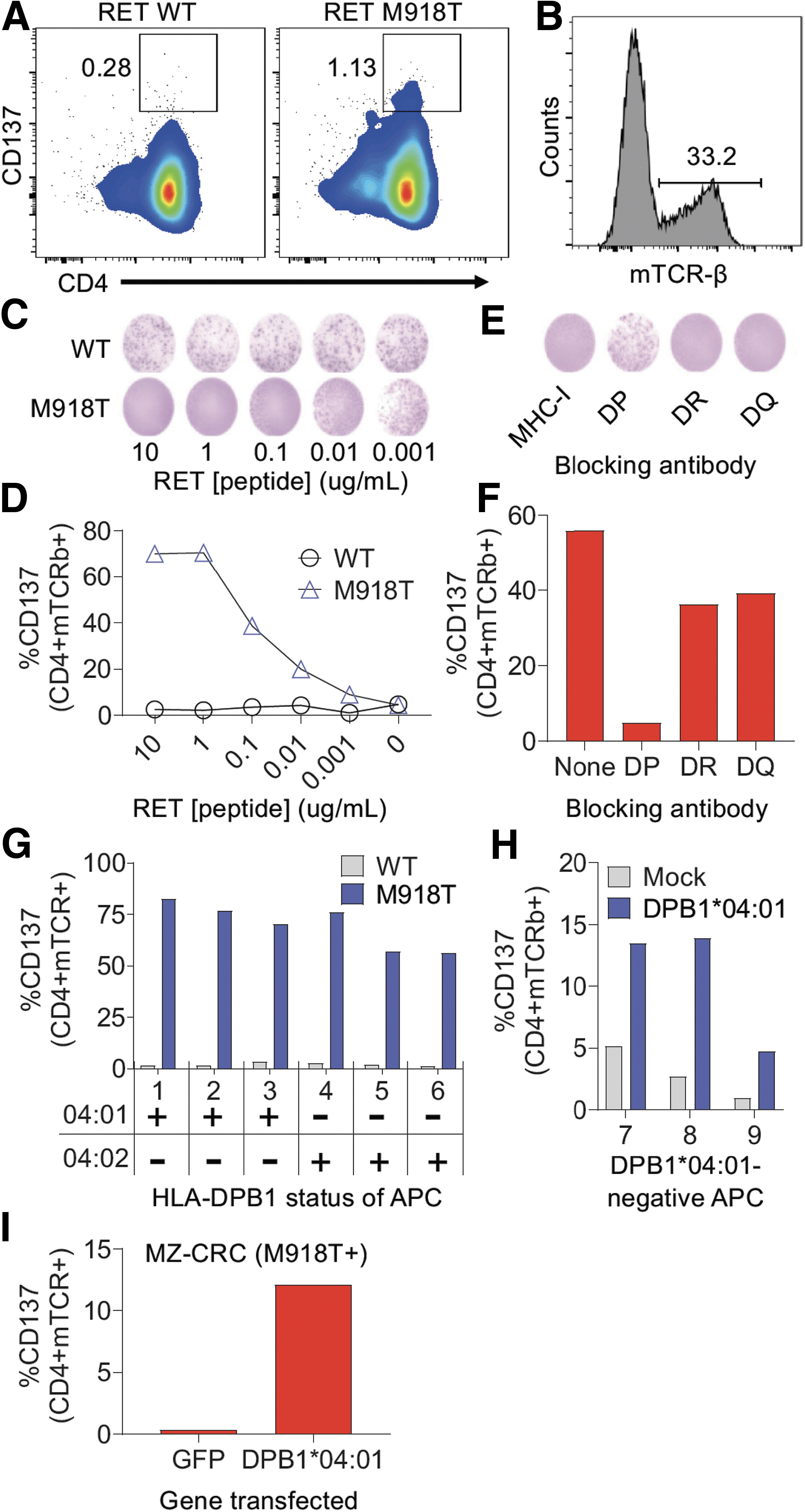
Identification and characterization of an MHC-II-restricted TCR targeting the RET M918T hotspot mutation in a patient with MTC. (
We used FACS to isolate the RET M918T-reactive T cells and sequenced the TCRs from these cells, which revealed a dominant TCR-α and TCR-β chain. To validate that this TCR indeed recognized RET M918T, we synthesized and cloned the genes encoding the TCRs into the MSGV1 retroviral vector and generated retroviral supernatants, which were then used to transduce autologous T cells (Fig. 4B). Coculture of the transduced T cells with autologous APCs pulsed with titrating doses of either WT or mutated M918T RET peptide demonstrated highly specific reactivity to RET M918T as determined by IFN-γ ELISPOT (Fig. 4C) and flow cytometric analysis of the T cell activation marker 4-1BB (Fig. 4D), with no reactivity against the WT RET peptide.
Identification of the HLA restriction element for RET-TCR
To determine which HLA class II molecule was presenting the RET M918T peptide to the CD4+ T cells, we performed coculture assays in the presence of blocking antibodies to the major HLA class II protein families HLA-DP, HLA-DQ, and HLA-DR, which revealed that HLA-DP was the HLA restricting element (Fig. 4E, F). Since the patient is homozygous at the HLA-DP locus, the RET M918T reactivity should be restricted by HLA-DPA1*01:03/HLA-DPB1*04:01 (hereafter referred to as “DPB1*04:01”), which is one of the most common HLA class II molecules in the human population.
To confirm this, we cocultured the transduced T cells with a panel of allogeneic APCs that either expressed DPB1*04:01 or the closely related DPB1*04:02, pulsed with the WT or M918T RET peptide and evaluated T cell reactivity the following day. As shown in Figure 4G, the transduced T cells recognized RET M918T presented by allogeneic APCs that expressed the DPB1*04:01 molecule or the DPB1*04:02 molecule. The transduced T cells displayed little reactivity to allogeneic APC that were mismatched at the HLA-DP locus unless the DPB1*04:01 gene was introduced by transfection (Fig. 4H).
Tumor cells process and present RET M918T on HLA-DP, which are recognized by RET-TCR
To determine whether the transduced T cells can recognize endogenously expressed RET M918T mutation (i.e., when the mutation is genetically expressed within a cell, in contrast to exogenously delivered as a peptide in the above experiments), the transduced T cells were cocultured with the MTC MZ-CRC cell line, which naturally expresses the RET M918T mutation, that was transfected with RNA encoding GFP or DPB1*04:01. As shown in Figure 4I, the TCR-transduced T cells only recognized the MZ-CRC cell line when DPB1*04:01 was transfected into the cell line. Thus, in a patient with MTC, we were able to identify and characterize a TCR derived from a CD4+ T cell that targets the RET M918T hotspot driver mutation when presented by the HLA-DPB1*04:01 molecule. Of note, we also found another HLA-DPB1*04:01-restricted RET M918T-reactive TCR from this patient, which appeared to have lower functional avidity than the RET-TCR described in Figure 4 (Supplementary Fig. S10).
Discussion
Despite recent advances in molecular targeted therapy and immunotherapy, metastatic MTC remains an incurable malignancy.
In terms of potential clinical translation, RET-TCR has the key advantage of targeting the tumor-specific RET M918T oncogenic driver mutation, which is only expressed by tumor cells and not in normal tissues and is therefore not expected to cause on-target/off-tumor toxicity. One important exception is in MEN2B patients who harbor the germline RET M918T mutation. 43 To the best of our knowledge, this is the first report of T cells targeting this MTC hotspot mutation. ACT using RET-TCR would be limited to patients who express the RET-M918T mutation, which is found in 30–40% of sporadic MTC patients 44 and who also express the HLA-DPA1*01:03/HLA-DPB1*04:01 allele combination (and potentially HLA-DPA1*01:03/HLA-DPB1*04:02), which are very common HLA-II allele combinations expressed by many U.S. Whites and Blacks. 45,46
Of note, the techniques described in this study could be used in other patients to determine whether there exists TCRs targeting RET-M918T in the context of other HLA alleles, or other recurrent RET mutations (e.g., RET C634), which if found, potentially could extend TCR gene therapy to additional patients with MTC. Although ACT has largely focused on harnessing cytotoxic CD8+ T cells, there is clinical evidence that transfer of CD4+ helper T cells can mediate regression of solid tumors 23,47,48 and thus RET-TCR may have therapeutic potential.
In contrast to neoantigen-specific RET-TCR, Cal24TCR, and MT-CAR target the tumor-associated self-antigens calcitonin and CEA, which are expressed by some normal tissues. The ultimate clinical utility depends on whether these self-antigens can be safely targeted without causing excessive or fatal on-target/off-tumor toxicity. Whereas TCR-transduced T cells targeting CEA induced partial tumor responses in colorectal cancer patients, they also caused severe side effects, including Grade 4 colitis. 29 However, CAR-T cells targeting CEA appear to have a more benign clinical safety profile relative to their TCR counterparts. 49,50 Notably, locoregional hepatic delivery of anti-CEA CAR-T cells were recently shown to induce a complete metabolic response of liver metastases in a pancreatic cancer patient without any major safety issues. 51
In contrast to CEA, calcitonin, which is also overexpressed in a subset of neuroendocrine lung and prostate tumors, 52,53 is a completely novel target that has not been targeted clinically with engineered T cell therapies. In cured thyroidectomized patients, calcitonin levels drop below the limit of detection (<2 pg/mL) indicating that the thyroid C cells are the body's major source of secreted calcitonin, and thus, the potential for autoimmunity might be limited to the thyroid, which is typically removed as first-line MTC treatment. 9 However, calcitonin is reported to be expressed in rare neuroendocrine cells in the lungs, 54 which may present a significant safety hurdle, as it remains to be seen how the potential loss of these rare cells would impact pulmonary function or if endogenous calcitonin expression levels are sufficient to confer recognition by Cal24TCR.
Furthermore, CALCA RNA is expressed in the liver, kidney, and prostate, per the GTEx database. 55 While targeting certain self-antigens (CD19, BCMA) with engineered T cells has proven highly effective, 26,27 the targeting of other self-antigens (HER2, MAGE-A3, MART-1) has resulted in severe toxicities, including therapy induced deaths, and thus, caution is warranted when targeting non-mutated self-antigens based on prior clinical experiences. 24,28,30
Our data indicate a lack of synergy for combining MT-CAR or Cal24TCR T cells in patients whose tumors are successfully controlled with RET kinase inhibitors, such as pralsetinib. Conversely, the emergence of RET solvent front mutations, 56 would likely lead to re-expression of high levels of calcitonin and CEA enabling recognition of RET inhibitor resistant clones by Cal24TCR or MT-CAR T cells.
Like other TCR-based therapies, the potential clinical utility of Cal24TCR is limited by HLA restriction and the potential loss of MHC observed in some tumors. Cal24TCR is restricted by HLA-A*24:02, which only is expressed in ∼15% of the U.S. population but is highly expressed in some Asian populations, such as over 76% of Taiwan's population. 45 However, an HLA-A2 calcitonin epitope has been reported, 57 and others are predicted based on IEDB screening, so it is conceivable that calcitonin-specific TCRs could be generated to cover a significant percentage of the population.
While MT-CAR is not subject to HLA restriction, clinical results for CAR-T cells thus far have been underwhelming in most solid tumors. 58 Unfortunately, xenogeneic NSG mouse models do not fully recapitulate tumor heterogeneity or the immunosuppressive tumor microenvironment T cells often encounter in human patients, as they lack human stromal components, vasculature, fibroblasts, Tregs, myeloid-derived suppressor cells (MDSCs), M2 macrophages, and other associated suppressive factors. 31 Moreover, normal tissue in NSG mice does not express human calcitonin, CEA, or HLA-A24, thus precluding evaluation of safety concerns such as on-target/off-tumor toxicity and cytokine release syndrome. As such, the potent in vivo efficacy results observed here should be interpreted in proper context.
To offset the potential toxicities associated with targeting self-antigens, additional genetic modifications, such as Syn-Notch receptors, 59 which might restrict expression of the CAR or TCR transgene to the tumor microenvironment could be employed. 60,61 Alternately, small doses of T cells could be injected locoregionally to limit off-tumor exposure. 49 Future work on Cal24TCR and MT-CAR should focus on novel methods to enhance tumor specificity.
In addition to potential safety concerns, the targeting of CEA and calcitonin may be limited by the non-essential nature of these proteins in the oncogenic process, which may lead to immune evasion due to outgrowth of antigen-low- or antigen-negative clones. In contrast, targeting RET M918T, which provides essential oncogenic signals, should be safe due to its tumor-specific expression. Providence Cancer Institute is in the process of opening a clinical trial to target hotspot mutations, including RET M918T, with TCR-gene therapy (NCT04520711). Overall, Cal24TCR, MT-CAR, and RET-TCR hold potential for use in generating cell therapies to treat metastatic MTC.
Footnotes
Authors' Contributions
T.A.E. designed the study and performed all experiments related to Cal24TCR and MT-CAR and wrote the corresponding sections. Y.-P.S. performed all experiments related to RET-TCR and wrote the corresponding sections. J.F. performed bioinformatics analyses. M.J. performed pralsetinib dose titration studies on the TT cell line. Studies were conducted in E.T.'s laboratory, and E.T. provided guidance and supervision throughout and editing of the article. All authors read and approved the article.
Acknowledgments
The authors would like to thank Dr. Jena French of the UC-Anschutz Medical Center for kindly providing the authenticated MZ-CRC-1 and TT MTC cell lines and ThyCa for providing generous financial support to fully cover in vivo study expenses. We also thank Miranda Gilchrist for assistance with fluorescence-activated cell sorting and Dr. Yaping Wu for support with IHC staining. We are grateful to the Providence Portland Medical Foundation (PPMF) and donors to the PPMF for funding parts of this study.
Author Disclosure Statement
E.T. is on the scientific advisory board of PACT Pharma, Genocea Biosciences, and Turnstone Biologics. All other authors report no disclosures or conflicts of interests.
Funding Information
This study was funded by the Providence Portland Medical Foundation and ThyCa.
Supplementary Material
Supplementary Methods S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12