
Abstract
Introduction:
Thyroblastoma, a primary thyroid neoplasm with histological features of primitive thyroid tissue has recently been described and is included as a distinct entity in the most recent edition of the World Health Organization (WHO) Classification of Tumors (5th edition). In this study, we expand the clinical, morphological, and molecular profile of this aggressive neoplasm.
Patient Findings:
The patients are females, 19 and 45 years of age, referred for large thyroid nodules. Tumor morphology is biphasic, composed of nests and follicles of epithelial cells, some with colloid-like secretions reminiscent of fetal thyroid follicles intertwined with a primitive stromal spindle cell component. By immunohistochemistry, the epithelial component is diffusely positive for PAX8 and TTF1 markers. Molecular studies showed DICER1 aberrations.
Conclusion:
A primary primitive thyroid malignancy reminiscent of early fetal embryology with no teratoid element, recently reported as thyroblastoma represents a unique entity, novel in its description, and is likely underdiagnosed.
Introduction
A wide variety of neoplasms histologically reminiscent of fetal, immature, or embryonal tissues have been described in the literature over the course of several decades. 1 –4 Although frequently encountered in the pediatric population, occasional cases have been reported in the adult population as well. 5 –8 In the thyroid, tumors with partially immature features, including spindle epithelial tumor with thymus-like differentiation, intrathyroid thymic carcinoma, and primary thyroid teratomas (TTs) are well documented. 9 –17
A primary thyroid malignancy with primitive thyroidal morphology, suggesting that the term thyroblastoma is more appropriate for what was called malignant teratoid tumor was recently reported, 18 and requires further substantiation as to warrant its recognition as an independent entity. The identification of somatic DICER1 alterations in thyroblastoma has led to the proposal that other malignant teratoid tumors, thyroid carcinosarcoma, and a subset of malignant TT may require reclassification. 17 –21 Distinguishing a thyroblastoma from other primary thyroid neoplasms with primordial features is crucial to precise tumor classification.
In this study, we expand on the clinical, morphological, and molecular profile of a recently identified entity with only primitive features reminiscent of fetal thyroid, best designated as thyroblastoma.
Case selection
We present two cases of thyroblastoma from our practice from 2016 to 2021. Patient demographics and clinical history were obtained from the medical record (Table 1). All available gross pictures, cytology slides, hematoxylin and eosin (H&E) stained slides, including ancillary studies and cytogenetic reports, were reviewed. For both cases, a thorough assessment was performed to identify and document all recognizable cytological and histological components.
Clinical and Pathological Features of Patients with Thyroblastoma
DOD, dead of disease; F, female; LN, lymph node; M, male; MNG, multinodular goiter; NA, not available; NED, free of disease; chemo, chemotherapy; TKI, tyrosine kinase inhibitor; XRT, external-beam radiation therapy.
Pathology findings
Both tumors had a fleshy tan-white surface on cut section with scattered hemorrhagic and necrotic foci. Both tumors were well circumscribed. Invasion into the adjacent normal thyroid parenchyma (Supplementary Fig. S1) was noted, but no extrathyroidal extension was observed.
Fine needle aspiration biopsy of lymph nodes was positive for neoplasia, including several microfollicles scattered in a background of lymphocytes. These microfollicles showed mild nuclear enlargement, crowding, and cytological atypia (Supplementary Fig. S2). PAX8 positivity in the microfollicles led to a diagnosis of metastatic thyroid malignancy.
Microscopic examinations of both cases revealed remarkably similar, almost overlapping morphological features. Both tumors showed an intrathyroidal nonencapsulated predominantly well-circumscribed tumor with several border irregularities, with invasion of the adjacent normal thyroid parenchyma. The tumor exhibited a biphasic growth pattern, composed of nests and cords of epithelial cells intermixed with stromal spindle cells (Fig. 1). The epithelial cells showed round and uniform nuclei with condensed to vesicular chromatin and scant to moderate amount of clear cytoplasm.

Hematoxylin and eosin–stained primary thyroid neoplasm with fetal-like features. (200 × ) The epithelial component shows a nested to tubular architecture closely admixed in a stromal/spindle cell background revealing a neoplastic biphasic nature.
In the second case, these epithelial areas showed occasional small central lumen with blastema-like tubule formation, some filled with a colloidal secretion, analogous to early fetal thyroid follicles. These primitive tubules were well observed in the first case with the nuance of being slightly better formed or more mature. No nuclear grooves or pseudoinclusions were observed. The spindled stromal cells have polygonal to elongated nuclei, vesicular chromatin, and moderate amounts of clear to eosinophilic cytoplasm (Supplementary Fig. S3).
Numerous mitoses averaging up to 20 mitoses per 10 hpf and scattered single-cell apoptoses were noted in both the epithelial and spindle cell stromal components of each case. Prominent vascular invasion was observed at the periphery of the tumor. Focal geographic necrosis was present. In the second case, histological assessment of the lung metastasis revealed the same biphasic morphology documented in the primary thyroid tumor, although some metastatic foci had rare minute areas of cartilage and were more spindle-cell component rich (Fig. 2). Thorough sampling of both tumors did not reveal any other ectopic embryonal components, such as immature cartilage, adnexal structures, pilosebaceous, and neuroectodermal tissue.
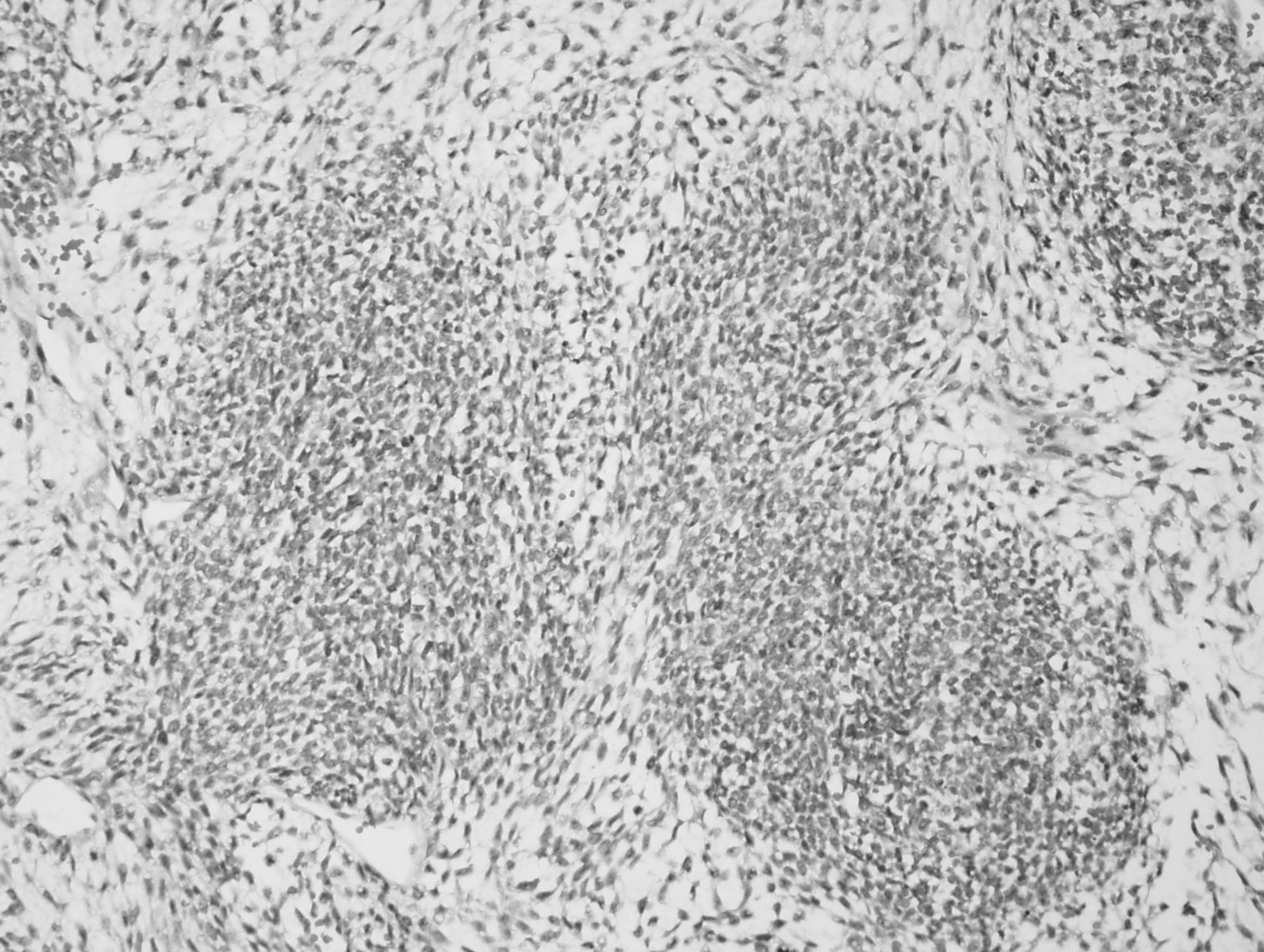
Hematoxylin and eosin–stained pulmonary metastasis by a biphasic epithelial-stromal tumor. (200 × ) Some metastases reveal a predominantly rich stromal component.
Immunohistochemical profile
Immunohistochemical studies were all executed on 5-μm sections of formalin-fixed paraffin-embedded tissue in a Bond 3 automated immunostainer (Leica Microsystems, Bannockburn, IL) and primary antibodies against several antigens were achieved (Supplementary Table S1). Appropriate positive and negative on-slide controls were included.
Both cases shared similarities in their immunohistochemical profiles. The epithelial component was diffusely positive for PAX8 (Supplementary Fig. S4A) and TTF1 (Supplementary Fig. S4B). The first case was negative for thyroglobulin, and the second showed rare, faint positivity (Supplementary Fig. S4C and Supplementary Table S1). In the spindle cell stromal component, the first case showed focal expression for p63 and retained wild-type p53. Spindle cells of the second case showed focal positivity for desmin, SMA, myogenin, and SALL4. The spindle cell stromal component of both cases was negative for PAX8, CD5, and neuroendocrine markers (Supplementary Table S1). The Ki67 proliferation index was over 60%.
Molecular analysis
Genotyping was performed using unstained sections from regions of interest identified by a board-certified pathologist after review of H&E-stained sections. After nucleic acid extraction, we performed next-generation sequencing. Specifically, we performed either (1) anchored multiplex PCR technology in combination with a high-throughput next-generation sequencing Illumina platform 22,23 or (2) a hybrid capture-based Food and Drug Administration (FDA) cleared next-generation sequencing assay (Personalized Genome Diagnostics, Baltimore, USA) (Supplementary Tables S2 and S3). All tests were performed in Clinical Laboratory Improvement Amendments (CLIA)-certified clinical laboratories. 22
Mutations typical of DICER1 syndrome were identified in both tumors. Genotyping in case 1 revealed a
For delineation of DICER1 mutation frequency we used a combination of computational queries. We used cBioPortal to obtain data from four large-scale thyroid cancer sequencing studies (The Cancer Genome Atlas [TCGA] Firehose, TCGA, Pancancer Atlas, and TCGA). 24,25 This was supplemented using data from the COSMIC databased subsetted for thyroid cancer (Supplementary Fig. S5 and Supplementary Table S4).
Electronic microscopy
Electronic microscopy was performed using formalin-fixed paraffin-embedded tissue of the main thyroid specimen. Selected tissue samples were processed using a standard protocol, as previously described. 26 All cases were analyzed by experienced electron microscopists (V.N. and M.S.). Images were captured with an advanced microscopy techniques digital charged-coupled device camera.
In the follicular areas, the presence of cuboidal cells containing small intracytoplasmic mitochondria and various amount of granular endoplasmic reticulum was noted. Some of these cuboidal cells were organized around small lumens and revealed small apical microvilli projecting into small- to moderate-sized colloid droplets (Supplementary Fig. S6). The spindle cells contained a moderate amount of cytoplasm with barely identifiable mitochondria and few distinguishing features. These cells were best described as primitive mesenchymal cells.
Discussion
Primary thyroid malignancies with a fetal/primitive morphology are unusual and likely under-recognized neoplasms. The thyroid is the earliest endocrine organs to differentiate and has an important hormonal role in embryonic development. By approximately day 24 of gestation, this anteriormost organ develops from a midline anlage swelling in the pharyngeal floor consisting of foregut endoderm cells. These thyroid precursor cells descend into the mesoderm above aortic sac and gradually forms into its mature shape with the isthmus connecting both lateral lobes. 27 By week 7 of gestation, the developing gland has reached its destination in the neck adjacent to the thyroid cartilage. 27
This pathway forms the typically temporary thyroglossal duct. The fetal thyroid reaches maturity by week 10–12 of gestation and nascent thyroid epithelial follicles start secreting colloid, the primary site of thyroid hormone production. Differentiated epithelial cells within these follicles, known as thyrocytes, possess an apical surface that delimits the follicle lumen and a basal (or basolateral) surface that faces the extrafollicular space. These cells primarily produce the thyroid hormone, thyroxine (T4) and triiodothyronine (T3, predominantly produced from T4 in the periphery) by week 16. 28 By that time, the fetal thyroid becomes active enough to support the fetal requirements for neural development. 28
The diagnoses of most cases classified as malignant teratoid tumor, malignant TT, and carcinosarcoma were initially used to refer to what it is presently better denominated as thyroblastoma. 18 –21 Their unusual morphological and immunohistochemical differences along with DICER1 alterations led to the classification of these tumors as a separate thyroid entity. 18 Thyroblastoma and TT share several morphological features and their distinction is essential. TT are germ cell neoplasms derived from all three germ cell layers: ectoderm, endoderm, and mesoderm. 17 TT can be separated into three categories based on their histological components: benign, immature, and malignant.
Histologically, TT is variable in lineage and percentages of diverse histotypes, including cutaneous adnexal structures, gastrointestinal, pancreatic, adipose, skeletal, cartilage, and glial tissues. Immature counterparts are composed of neuroepithelium, neuroblastemal elements, and embryonic mesenchymal connective tissue. Embryonic mesenchymal tissue can easily be recognized by immunohistochemistry as it expresses desmin and occasionally MyoD in areas of early rhabdomyoblastic differentiation. 19,29 Molecular characterization of these neoplasms has revealed gain of chromosome 12, and loss of 17p. 19,30
In previous studies, thyroblastoma showed a morphological triad composed of solid nests composed of primitive small round cells, primitive appearing thyroid follicles, and fetal-type glands. These structures were embedded in a spindle cell stroma with occasional rhabdomyoblastic or sarcoma-like differentiation. 18 –21 Few cases had heterologous elements of cartilage present. 18,19 Our cases only revealed primitive appearing thyroid follicles and primitive tubules or gland-like arrangements. The proportion of epithelial and stromal components varied in each tumor and metastasis, which suggests for a wider spectrum of histological features in thyroblastoma. The morphological triad is not necessary for the diagnosis.
All reported cases including ours revealed DICER1 anomalies. 18 –21 Mutations in DICER1 gene may be of somatic or germline origin. In regard to somatic mutation in thyroid neoplasms, a spectrum of DICER1 alterations has been identified within adenomatous nodules, follicular hyperplasia, papillary thyroid carcinomas, follicular thyroid carcinomas, poorly differentiated thyroid carcinomas of childhood, anaplastic thyroid carcinomas, and thyroblastoma. 18 –21,24,25 In thyroblastoma, DICER1 anomalies does not seem to have a specific phenotype–genotype.
DICER1 syndrome is an inherited pleiotropic tumor predisposition syndrome characterized by a distinctive constellation of neoplastic and dysplastic lesions primarily affecting children and young adults. The syndrome is caused by germline pathogenic variants in the DICER1 inherited in an autosomal dominant pattern. 31 –35 Most patients with DICER1-related thyroid disease present with multinodular thyroid disease, including multinodular hyperplasia and multiple adenomatous nodules. The presence of intrafollicular centripetal papillary growth is a characteristic finding in this syndrome. 18,34 There are also involutional changes in the non-nodular thyroid parenchyma in DICER1-related thyroid disease.
The tumors described in DICER1 syndrome are the low-risk follicular-patterned differentiated thyroid carcinoma, including follicular thyroid carcinoma and papillary thyroid carcinoma. 34,35 Patients with this syndrome may present with other tumors, including renal pediatric-type cystic nephroma, pleuropulmonary blastoma, sinonasal chondromyxoid hamartomas, gonadal sex cord stromal tumors, Mullerian adenosarcoma, pituitary blastoma, pineoblastoma, PNET, and other sarcomas. 31 –35 Thyroid tumors are now recognized to be associated with somatic DICER1 mutations, and DICER1 mutation identified in our both cases were of somatic origin. Our patients had no past medical of other tumors or familial history of any other tumor or syndrome.
Unusual and morphologically complex neoplasms are rare but certainly encountered with some regularity in the thyroid gland. The cases detailed in this study have some unique features encountered in the tumor types described herein. Particularly, the tumors we described exhibited some architectural and immunohistochemical features seen among rare thyroid entities previously described; however, it is our opinion that these shared features were insufficient to appropriately classify our cases. In our presented unique cases, the tumors reveal an immunohistochemical profile consistent with solely thyroidal differentiation; more specifically, thyroid follicular cell lineage. Importantly, the tumor morphology is reminiscent of early fetal thyroid tissue, thyroblasts.
There was no evidence of any other components, such as mature or immature neural, pilosebaceous, cartilage, or adnexal structures identified that could suggest an underlying malignant teratoma. An additional challenge in the diagnosis of teratomas may lie in the interpretation of the 2016 World Health Organization (WHO) Classification of Endocrine and Neuroendocrine Tumors, which seems to require all three germ cell layers to be present for the diagnosis to be granted. This definition differs from teratomas of the female reproductive organ or other organs, as only two germ cell layers are needed. It is our opinion that neoplasms with a fetal-like/primitive thyroid morphology, a positive thyroid immunohistochemical profile, and somatic DICER1 anomalies represent the same tumor subtype and should be classified distinctly as a thyroblastoma, an embryonal high-grade thyroid neoplasm.
Footnotes
Authors' Contributions
J.G. wrote, reviewed, and designed the article; collected, contributed, and analyzed data. D.D.-S., M.S., and D.A.H. contributed and analyzed data. J.L. reviewed the article, and contributed and analyzed data. P.M.S. reviewed the article and analyzed data. V.N. wrote, reviewed, and designed the article; identified the tumor types; collected, contributed, and analyzed data.
Ethical Approval
The authors are accountable for all aspects of the study in ensuring that questions related to the accuracy or integrity of any part of the study are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was approved by the Mass General Brigham Institutional Review Board (2012P001024, P.M.S. PI) and individual consent for this retrospective analysis was waived.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This article was funded by intradepartmental funding at Massachusetts General Hospital.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4