
Abstract
Background:
Anaplastic thyroid cancer (ATC) is an aggressive solid cancer in humans with few treatment options. Recent studies suggest that aberrant gene transcription could contribute to aggressive ATC progression. To test this hypothesis, we assessed if blocking cyclin-dependent protein 7 (CDK7) activity could impede ATC progression through attenuation of cancer stem cell (CSC) activity.
Methods:
We treated cell lines isolated from human ATC (THJ-11T and -16T) and xenograft mice induced by these cells with the CDK7 inhibitor THZ1. Through integrative transcriptome analyses we found that the NOTCH1-cMYC signaling axis was a potential target of CDK7 inhibition in ATC. To determine the regulatory action of NOTCH1-cMYC signaling in CSC maintenance, we evaluated the effect of a selective NOTCH1 inhibitor, crenigacestat, on CSC capacities in ATC.
Results:
THZ1 markedly inhibited proliferation of ATC cells and xenograft tumor growth by blocking cell cycle progression and inducing apoptosis. NOTCH1 was sensitive to suppressive transcription mediated by CDK7 inhibition and was highly enriched in tumorspheres from ATC cells. Treatment of ATC cells with either crenigacestat or THZ1 blocked formation of tumorspheres, decreased aldehyde dehydrogenase activity, and suppressed in vivo initiation and growth of tumors induced by ATC cells, indicating that NOTCH1 was a critical regulator of CSC activity in ATC. Furthermore, we demonstrated that cMYC was a downstream target of NOTCH1 signaling that collaboratively maintained CSC activity in ATC. Of note, genomic analysis showed that low CDK7 expression contributed to longer disease-free survival of thyroid cancer patients.
Conclusions:
NOTCH1 is a newly identified CSC regulator. Targeting NOTCH1-cMYC signaling is a promising therapeutic strategy for ATC.
Introduction
Anaplastic thyroid carcinoma (ATC) is one of the most aggressive solid tumors afflicting humans. Although ATC is rare, encompassing 1–2% of all thyroid cancers, this cancer causes more than half of all thyroid cancer-associated deaths. 1,2 In most cases, differentiated thyroid cancer (DTC) is treated through a combination of surgery, adjuvant radioactive iodine ablation, and thyrotropin-suppressive therapy resulting in high recovery rates, even in cases of extrathyroidal tumor manifestation. However, the more extensive spread of ATC often precludes complete resection and the absence of sodium iodine symporter expression excluded the use of radioiodine treatment. Patients with metastatic ATC rarely survive more than a year after diagnosis.3
High mortality rates are driven by the paucity of effective treatments or viable strategies to slow or mitigate cancer progression. Crucially, development of effective treatments is hampered by a relatively poor understanding of the genetic basis underlying the dedifferentiation process and the progressive genetic changes occurring during ATC carcinogenesis. However, recent application of comprehensive next-generation sequencing (NGS) analyses has provided novel insights on molecular and genetic changes associated with the complex genomic and transcriptomic landscape of ATC carcinogenesis. 4 –7 These include identification of mutated driver genes involved in thyroid carcinogenesis and transcription, such as SWI/SNF subunit genes and EIF1AX, as well as chromatin modifiers such as histone methyltransferases (KMT2A, KMT2C, KMT2D, and SETD2) that were mutated in ATC. 4,5 Together, these observations suggest that transcriptional regulation could have a critical role in anaplastic manifestations of ATC.
However, the mechanisms by which abnormal transcription factors impact ATC development remained unclear. We therefore adopted an approach to understand how loss of RNA polymerase activity, the central player of transcription, affects development of ATC. The key to our approach was THZ1, a specific inhibitor of cyclin-dependent protein 7 (CDK7). CDK7 is a member of the serine/threonine protein kinase family that is critical for maintenance of proper regulation of gene transcription. CDK7 regulates mRNA transcription initiation and elongation by phosphorylation of Ser-2 and Ser-5 of the carboxyterminal domain (CTD) of RNA polymerase II (RNAPII). 8 –10 THZ1 binds to CDK7 covalently at a cysteine residue, impairing the ability of CDK7 to effectively phosphorylate RNAPII. THZ1 was recently shown to inhibit expression of genes involved in cell proliferation and suppression of apoptosis in several cancers. 11 –14
Using THZ1 as a probe, we found that THZ1 effectively inhibited the phosphorylation of CTD of RNAPII in two cell lines (THJ-11T and -16T) isolated from ATC patients. We also found that THZ1 suppressed cell proliferation and induced apoptosis of ATC cells in vitro and within in vivo xenograft tumors. We further demonstrated that suppression of tumor growth by THZ1 was caused by the blocking of cancer stem cell (CSC) activity and was mediated by inhibition of the NOTCH1-cMYC transcription network. Our results provide new insights into the critical involvement of transcription along the NOTCH1-cMYC axis in the carcinogenesis of ATC.
Materials and Methods
Transcriptome data analysis
We analyzed transcriptome data from 505 thyroid cancer samples and 59 normal samples provided by The Cancer Genome Atlas-Thyroid Cancer (TCGA-THCA;
To assess possible association of the NOTCH1-cMYC signaling axis with observed CSC activity, we used a machine-learning algorithm to estimate a “Stemness Index” that reflects relative levels of TCGA-THCA.16 To identify gene signatures underlying the biological changes by THZ1 treatment or associations between two signaling pathways, we performed gene-set enrichment analyses (GSEA;
Cell culture
ATC cell lines (THJ-11T, -16T, -21T, and -29T) and normal thyroid cell lines (THJ-116N and -101N) were provided by Dr. John A. Copland, III at the Mayo Foundation for Medical Education and Research.17 FTC-133 and FTC-236 cells derived from follicular thyroid carcinoma, were kindly provided by Dr. Orlo H. Clark (UCSF Mount Zion Medical Center, San Francisco, CA, USA). The authentication of cell lines was validated by short-tandem DNA repeat in a previous study.17 Further details are given in Supplemental Information.
In vivo mouse xenograft study
For all animal experiments in this study, we followed protocols approved by the National Cancer Institute Animal Care and Use Committee. We used about six-week-old female athymic nude mice as previously described.18 The treatment group received an intraperitoneal injection of THZ1, 10 mg/kg twice a day intraperitoneally, for 2 weeks, whereas the control group received vehicle solution. To assess the tumor-initiating capacity, we performed in vivo limiting dilution assay as previously described.19 We followed the ARRIVE Guidelines 2.0 for reporting in vivo experiments. Further details are given in Supplementary Information.
Tumorsphere formation assay
ATC cells (11T and 16T cells) (2 × 103 cells) were plated in 96-well low attachment plates (Corning) containing media treated with either THZ1, crenigacestat, or dimethyl sulfoxide (DMSO) as described previously.18 The number and size of tumor spheres were measured and photographed by a Zeiss light microscope after 5–7 days.
Flow cytometry analysis (fluorescent-activated cell sorting)
ATC cells (11T and 16T cells) were treated with the indicated dose of THZ1, crenigacestat, or DMSO for 24 or 48 hours, harvested, and subject to each assay as described previously.18
Statistical analyses
Continuous variables were compared using a two-tailed Student's t-test, and categorical data were compared using a two-tailed chi-squared test or Fisher's exact test. Pearson correlation coefficients were used for the correlation analyses of cMYC, NOTCH1, and CDK7 gene expressions. To diagram the results from these analyses, we used the RStudio 1.2 program and GraphPad Prism software. Two-way analysis of variance with Bonferroni's post hoc tests were applied to compare tumor growth rates in the xenograft model according to the time and different groups. Disease-free survival was compared between low and high CDK7 groups using a Kaplan–Meier estimator. Data are presented as mean ± standard deviation, and we used SPSS version 23.0 or GraphPad Prism for statistical analyses. All p-values were two-sided throughout, and those <0.05 were considered statistically significant.
Additional methods are given in Supplemental Information.
Results
Blocking of CDK7 activity greatly inhibits ATC cell proliferation
We first used TCGA-THCA database to analyze the association of CDK7 expression patterns with degree of aggressiveness of thyroid cancer. Expression of CDK7 mRNA increased during oncogenesis and dedifferentiation of thyroid cancer (Fig. 1A) and had a strong negative association with thyroid differentiation scores (Fig. 1B). This pattern was consistent with trends of CDK7 protein levels in thyroid cancer cells with different degrees of differentiation. CDK7 protein abundance (Fig. 1C) was highest in the four highly dedifferentiated ATC cell lines (29T, 21T, 16T, and 11T cells) compared with normal thyroid cells (116N and 101N) and with differentiated follicular thyroid cancer cells (FTC236 and FTC133). The elevated CDK7 protein levels were accompanied by increased total RPB1 CTD levels (both unphosphorylated and phosphorylated forms) (Fig. 1C), the largest subunit of RNAPII. RPB1 CTD was detected by the antibody (Supplementary Table S3), widely used by other investigators to detect total RPB1 CTD abundance. 20 –22 These findings indicated that active transcription was associated with thyroid cancer progression into ATC.
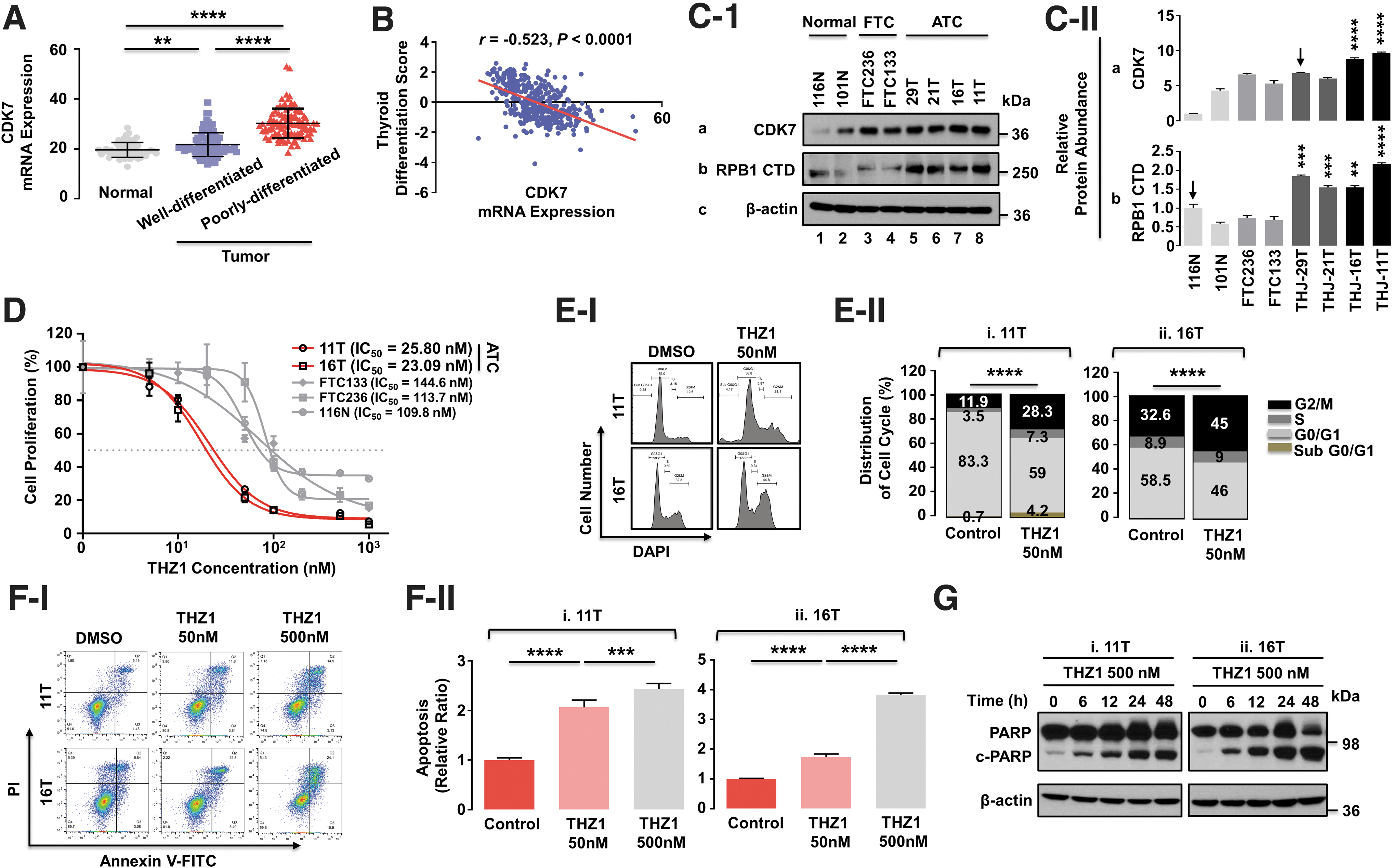
Suppression of CDK7 activity by THZ1 inhibited cell proliferation via blocking of cell cycle progression and induction of apoptosis. (
The relatively high expression of CDK7 in the ATC cell lines enabled exploration of the consequences of suppressive transcription mediated by CDK7 inhibition in ATC progression. We therefore tested effectiveness and selectivity of inhibitory effects of THZ1 on CDK7 in thyroid cancer cells. The 11T and 16T cells, which had high CDK7 protein levels, were more sensitive than FTC133, FTC236, and 116N cells to the inhibitory effects of THZ1 (Fig. 1D).
During cell cycles and apoptosis where THZ1 inhibited the proliferation of ATC cells, THZ1 markedly blocked cell cycle progression through the G2/M phase, as 11T and 16T cells at G2/M increased from 11.9% to 28.3% and from 32.6% to 45%, respectively (Fig. 1E). THZ1 dose dependently increased annexin V-positive apoptotic cells (Fig. 1F) and cleavage of poly (ADP-ribose) polymerase (c-PARP) (Fig. 1G) in both ATC cells. Together, these results are evidence that (a) THZ1 effectively inhibits ATC cell proliferation by blocking cells from transiting through the G2/M phase and by induction of apoptosis, and (b) that suppression of RNAPII transcription activity via CDK7 inhibition impedes ATC progression.
Inhibition of CDK7 activity by THZ1 suppresses ATC cell-induced tumor growth
Evidence that THZ1 strongly inhibited proliferation of ATC cells in vitro prompted us to test effects of THZ1 on xenograft tumor growth. Treatment of nude mice with THZ1 suppressed growth of tumors induced by 11T cells (n = 9; Fig. 2A-I and II) and by 16T cells (n = 10; Fig. 2B-I and II) as THZ1 treatment led to a 40% reduction in 11T tumor weights (Fig. 2A-III) and a 50% weight reduction in 16T tumors (Fig. 2B-III). Immunohistochemical analyses (Fig. 2C, D) consistently showed that THZ1 inhibited the proliferation of tumor cells, as evident from reduced Ki-67 positively stained ATC cells, by decreasing cyclin D1 levels, and inducing apoptosis as evident from elevated cleaved caspase-3 expression. Together, these in vivo results indicate that inhibition of CDK7 activity by THZ1 is a potentially viable approach to treatment of ATC patients.
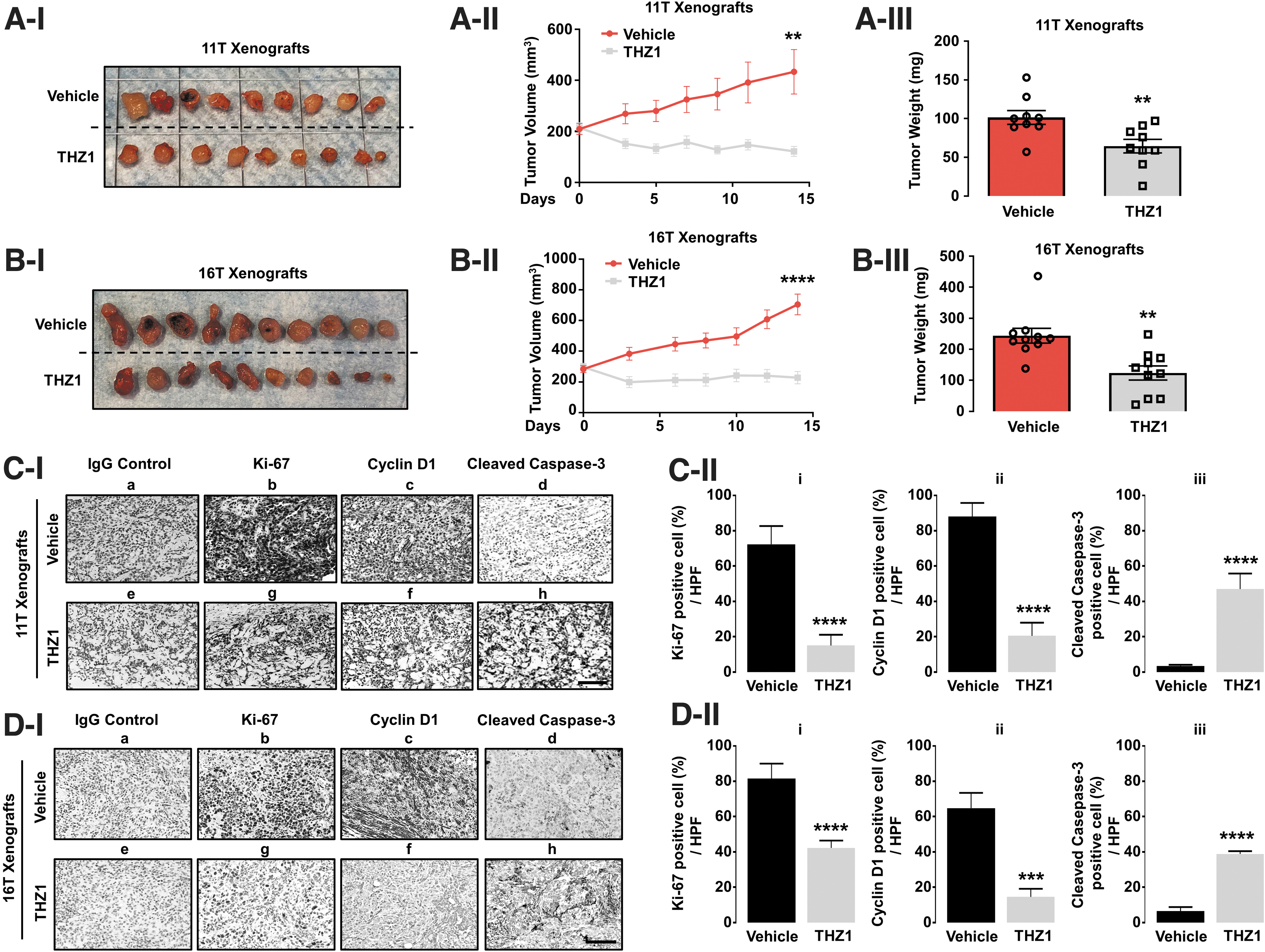
THZ1 inhibits growth of tumors induced by 11T or 16T cells in mouse models. (
Suppression of NOTCH1 expression by THZ1 blocks CSC activity in ATC
To identify additional candidate genes regulated by CDK7 activity, we searched downregulated gene sets in THZ1-treated cells using gene set enrichment analyses of public dataset GSE116282. Among the downregulated genes, NOTCH1 signaling was notable (Supplementary Fig. S1).23 Subsequent analyses (Supplementary Fig. S2) showed that NOTCH1 mRNA expression was suppressed by 80% in esophageal,24 nasopharyngeal,25 breast,26 and ovarian cancer cells23 by THZ1.
We next compared NOTCH1 mRNA expression among normal thyroid, papillary thyroid cancer (PTC), and ATC, which had the highest expression of NOTCH1 (Fig. 3A). We next examined how NOTCH1 was linked to CDK7 activity at the transcription level. In both 11T and 16T cells, THZ1 suppressed protein levels of phosphorylated RNAPII CTD at serine sites (Ser-2, 5, and 7; Fig. 3B). Of note, RPB1 CTD was suppressed in as little as 6 hours and for as long as 48 hours. In addition to the virtually total suppression of these transcription regulators, no NOTCH1 protein was detectable in either 11T or 16T (Fig. 3B). Similarly, the expression of the ligands for NOTCH1 receptors, including delta-like 1 (DLL1), and Jagged1 (JAG1) were inhibited by THZ1 treatment in both ATC cells (Fig. 3C). The functional consequences of suppressing expression of NOTCH1 receptors and ligands by THZ1 were evident from similar decreases in expression patterns of downstream target genes HES1, HEY1, MAML1, and EP300 (Fig. 3C).
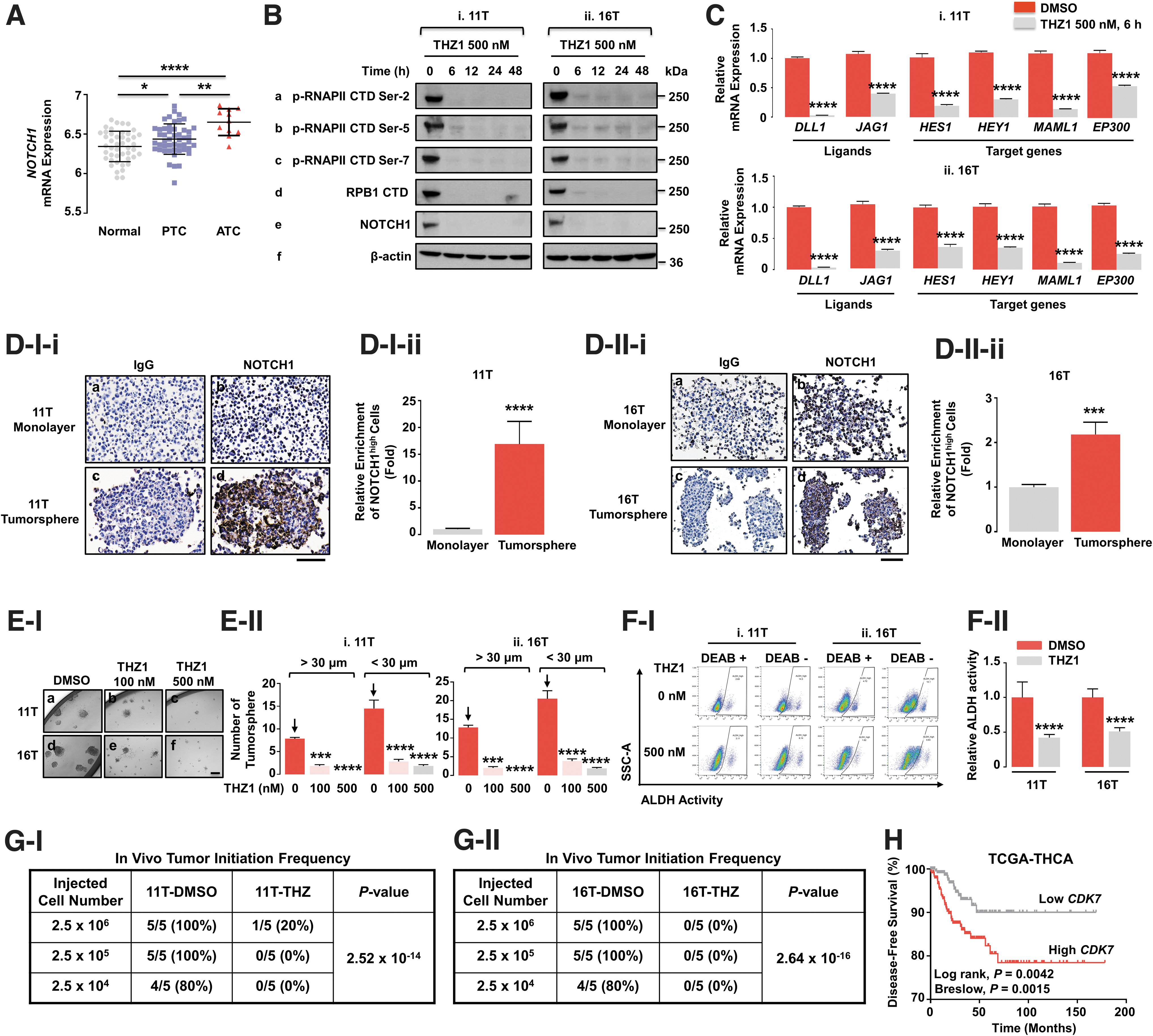
NOTCH1 signaling is critical for CSC maintenance in ATC. (
The observation that NOTCH1 receptors could contribute to CSC activity in ATC tumorigenesis was examined further using immunohistochemical and fluorescent-activated cell sorting (FACS) analyses. Figure 3D-I shows that stronger staining of NOTCH1 was detected in tumorspheres of 11T cells compared with those cultured in the monolayers. Of note, an almost ninefold larger proportion of NOTCH1 proteins was concentrated in the tumorspheres (Fig. 3D-I-ii). Similar elevated NOTCH1 protein concentrations were documented in tumorspheres of 16T cells compared with monolayer cells (Fig. 3D-II). These findings indicate highly enriched levels of NOTCH1 proteins are associated with CSC.
The THZ1 inhibition of NOTCH1 protein levels given in Figure 3B is consistent with hypothesized suppression of CSC activity by THZ1. Indeed, we found that THZ1 at 100 nM partially suppressed tumorsphere formation of 11T and 16T cells, but at 500 nM, THZ1 almost totally blocked tumorsphere formation (Fig. 3E-I). Quantitative analyses showed that both small (<30 μm) and large (>30 μm) tumorspheres were reduced by THZ1 treatment (Fig. 3E-II). The suppression of CSC activity by THZ1 was also supported by 50–60% inhibition of aldehyde dehydrogenase (ALDH) activity in both ATC cells shown by FACS analysis (Fig. 3F). The in vitro inhibition of CSC activity by THZ1 was further confirmed by in vivo limiting dilution analysis (Fig. 3G and Supplementary Table S1). Of interest, pretreatment with THZ1 almost completely blocked tumor initiation of 11T and 16T cells (Fig. 3G-I and II, respectively). Analysis of TCGA-THCA database showed that low CDK7 expression was associated with more prolonged disease-free survival of thyroid cancer patients (Fig. 3H). Together, these results provided strong evidence that the inhibition of CDK7 activity by THZ1 blocks CSC at least, in part, by suppressing NOTCH1 signaling, to potentially prevent tumor recurrence in humans.
NOTCH1-cMYC signaling axis is critical for maintaining CSC activity in ATC
Our finding that NOTCH1 is a regulator of CSC activity in human ATC prompted us to identify downstream regulators of NOTCH1 signaling in ATC. Previous studies have shown that cMYC is a NOTCH1 transcription target in breast cancer27 or T cell acute lymphoblastic leukemia/lymphoma. 28,29 However, the possibility that cMYC is functionally linked to NOTCH1 signaling in ATC was not clear. Comparison of mRNA expression of cMYC showed that ATC had markedly higher expression levels than that of normal thyroids and PTC (Fig. 4A). In addition, there was a strong positive correlation of cMYC and CDK7 expression levels in ATC, but not in the normal thyroid and PTC (Fig. 4B), suggesting that the expression of cMYC transcriptional activity could also be sensitive to THZ1. This suggestion was further supported by coordinated enrichment of cMYC target genes through high CDK7 expression in humans (Supplementary Fig. S3). We therefore tested the effects of THZ1 in 11T and 16T cells with elevated cMYC protein levels (Fig. 4C), compared with the normal and follicular thyroid cancer cells. In both ATC cells, the THZ1 treatments led to reduced cMYC protein levels and phosphorylation of RNAPII CTD in concentration-dependent manner (Fig. 4D-I and -II). In summary, THZ1 treatment blocked expression of NOTCH1 (Fig. 3B) and cMYC in ATC while concomitantly suppressing RNAPII activity.
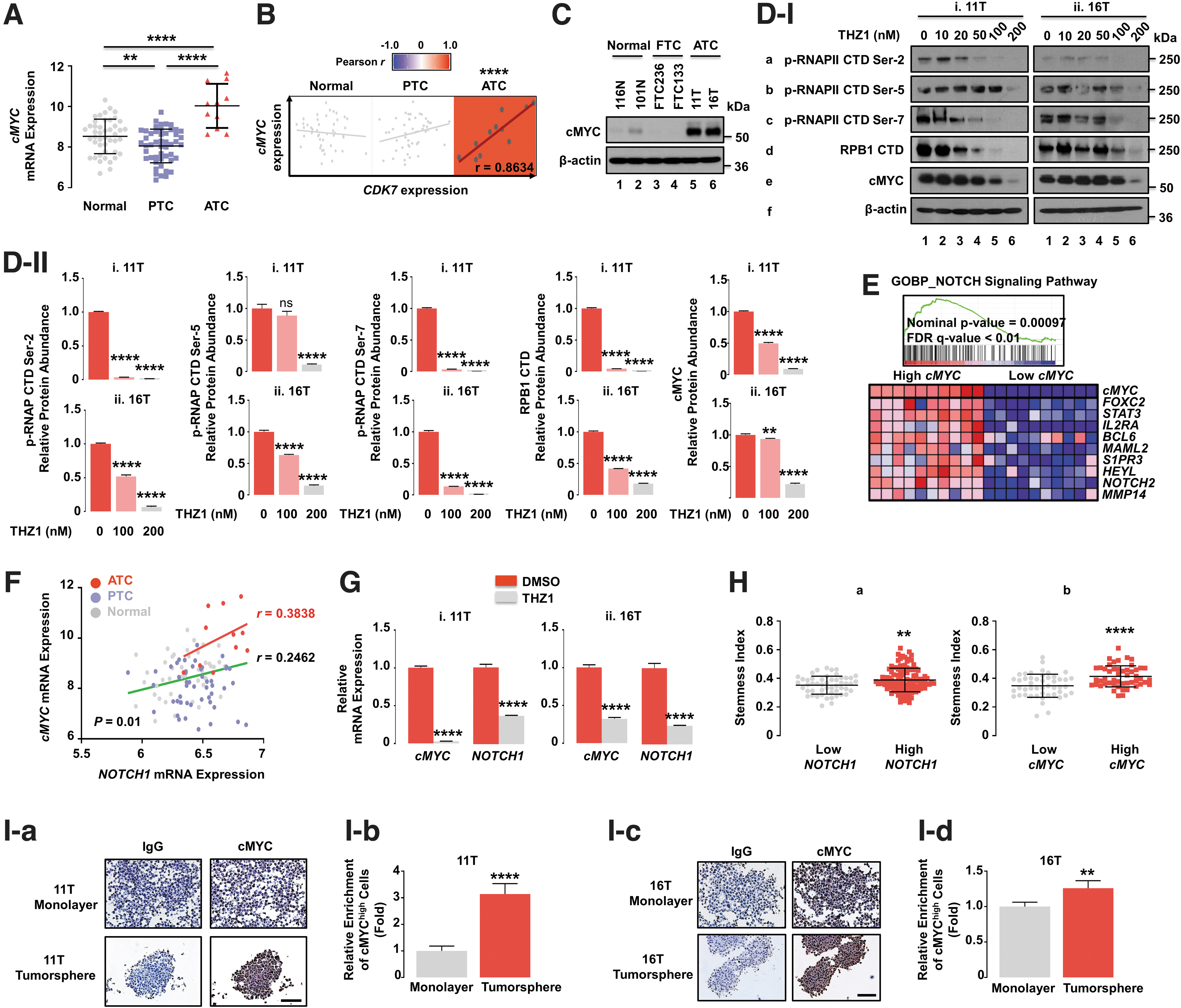
Functional link of NOTCH1 to cMYC in maintaining cancer stemness. (
We next conducted gene set enrichment analysis to show that the NOTCH1 signaling pathway was highly correlated with cMYC mRNA expression (Fig. 4E). The functional link of NOTCH1 with cMYC signaling was further demonstrated by observations of a stronger positive correlation of mRNA expression of these two genes in human ATC compared with normal thyroid and PTC (Fig. 4F), suggesting that NOTCH1-cMYC is a critical signaling axis for thyroid cancer progression into ATC. This correlation was further supported by showing that THZ1 treatment led to concurrent repression of mRNA expression of both NOTCH1 and cMYC genes in ATC cells (Fig. 4G). Furthermore, we found similar close associations of high stemness with elevated NOTCH1 and cMYC expression in human thyroid cancer (Fig. 4H), which was validated by elevated cMYC expression in the tumorspheres of ATC cells (Fig. 4I), as given in Figure 3D for NOTCH1. These observations further supported a link of NOTCH1 with cMYC signaling in ATC.
We next demonstrated the association of NOTCH1 with cMYC in xenograft tumors (Fig. 5A, B). We first showed that THZ1 was effective in suppressing the transcription machinery by reducing phosphorylation of RNAPII CTD Ser-2 and Ser-5 in 11T and 16T-induced tumors. This transcriptional suppression by THZ1 led to a dramatic and coordinated reduction in protein expression of the CSC marker, ALDH, NOTCH1, and cMYC. These findings suggested NOTCH1-cMYC functionally linked to CSC activity in ATC, which was crucial for tumor growth (Fig. 2).
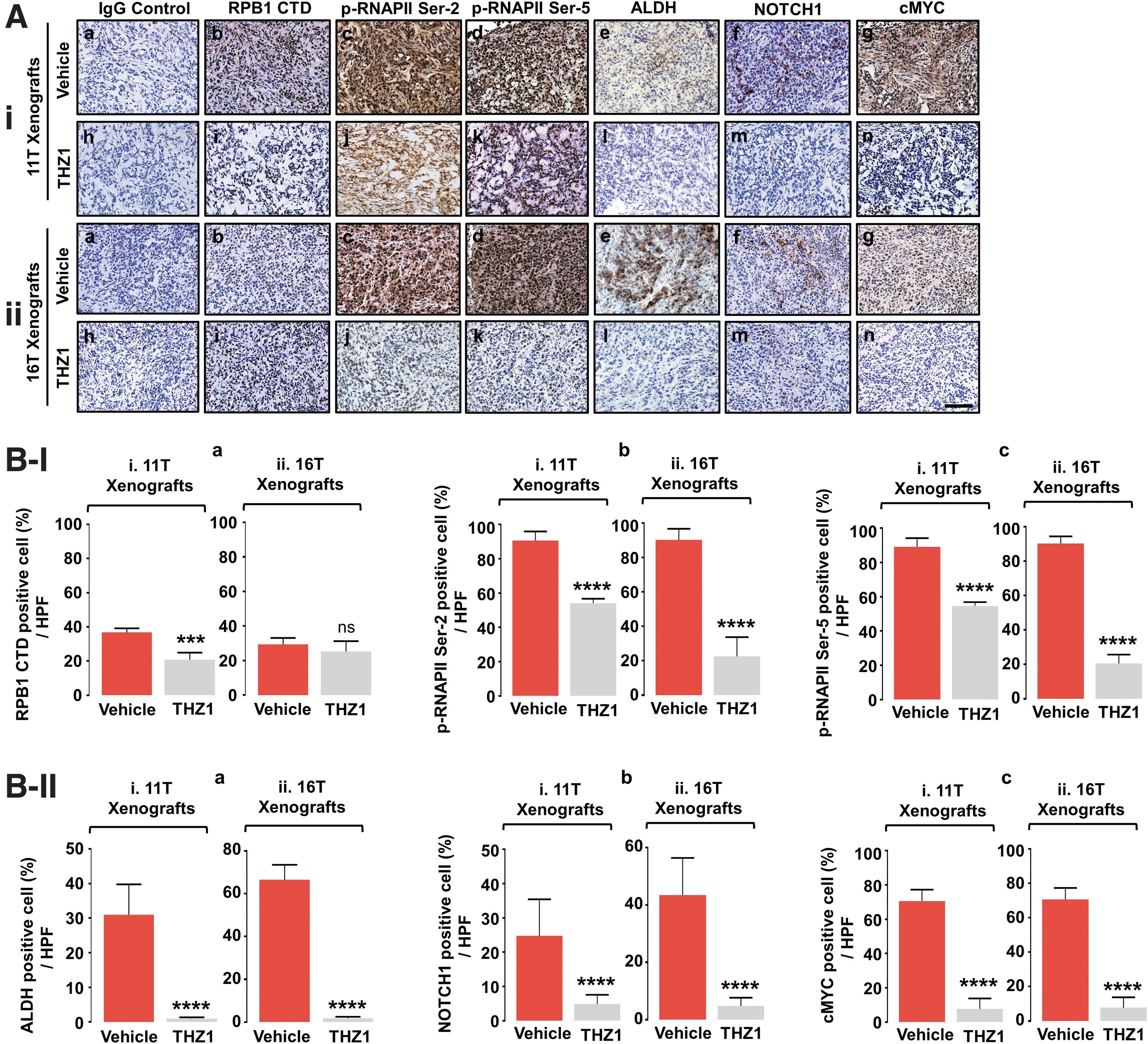
Concurrent transcription suppression of NOTCH1 and cMYC genes via inhibition of CDK7 by THZ1 in vivo. (
To show that cMYC is a direct target of NOTCH1 signaling in ATC, we assessed whether inhibition of NOTCH1 activity by crenigacestat could attenuate cMYC activity. Crenigacestat, a potent small-molecule inhibitor of proteolytic γ-secretase activity, can prevent release of the NOTCH intracellular domain (NICD), thereby modulating NOTCH1 signaling and its downstream biological effects.30 The effectiveness of crenigacestat in inhibiting NOTCH1 signaling was apparent from the reduction in protein levels of NICD, the active form of NOTCH1, without affecting the transmembrane domain (NTM) and full-length NOTCH1 in ATC cells (Fig. 6A-I). Of importance, cMYC protein levels were correspondingly lower (Fig. 6A-I and -II), establishing that cMYC is the downstream target of NOTCH1 signaling in ATC.

NOTCH1 is a newly identified CSC regulator to promote tumor initiation and progression in ATC. (
The functional consequences of suppression of NOTCH1-cMYC signaling in ATC were evident from the near total blockage of larger (>30 μm) tumorsphere formations (Fig. 6B, C) and the strong suppression of ALDH activity (Fig. 6D, E) by crenigacestat treatment. We subsequently assessed the relative significance of NOTCH1-cMYC on CSC activity in vivo with an in vivo limiting dilution assay. In DMSO-treated 11T cells, matching numbers of tumors were induced for each cell number inoculated. As expected, the tumor size progressively decreased as fewer cells were inoculated. For each treatment (number of cells), crenigacestat markedly suppressed tumor initiation and growth (Fig. 6F). At the lowest cell number (2.5 × 104 cells), only one of five mice injected with crenigacestat-pretreated 11T cells developed a (small) tumor, while four of five mice injected with DMSO-pretreated 11T cells developed larger tumors, which was reflected by a 90% reduction of CSC frequency by crenigacestat (Supplementary Table S2). The suppression of tumor growth by crenigacestat was also reflected in the growth curves (Fig. 6F-II), providing further evidence of the essential role of CSC maintenance through NOTCH1 signaling for both ATC initiation and progression. This finding was further supported by a 70% reduction of tumor weight by crenigacestat (Fig. 6F-III) and by similar inhibitory effects on growth of tumors induced by 16T cells pretreated with crenigacestat (Fig. 6G). Broadly, the NOTCH1-cMYC signaling axis was critical for ATC initiation and progression by maintaining CSC activity.
Discussion
Treatment of de-DTC, including ATC, has evolved from a reliance on chemotherapy to a more recent phase of targeting tyrosine kinases. However, the long-term efficacy of tyrosine kinase inhibitors has been limited by the development of resistance to the activity of specific kinase inhibitors in patients. However, the advent of powerful NGS approaches has uncovered previously unrecognized molecular events underlying carcinogenesis at genetic and genomic levels. One striking outcome of such approaches was the discovery of the numerous relatively common mutations in the various components of the transcription machinery, including the SWI/SNF chromatic remodeling complex and methyltransferase epigenic modifiers in ATC.4 These findings suggested that aberrant regulation of transcription perhaps contributes to progression of ATC. 18,31,32
Using a CDK7 inhibitor to probe the potential impact of aberrant transcription on ATC carcinogenesis, we found that NOTCH1 expression was sensitive to the effects of THZ1. While NOTCH1 has been reported to be critical for breast cancer tumorigenesis,27 leukemia, and lymphomas, 28,29,33 its role in ATC was less known. We report here that NOTCH1 is highly enriched in tumorspheres of 11T and 16T cells, and that treatment of these cells with THZ1, through significant inhibition of NOTCH1 transcription, disrupted the formation of tumorspheres and suppressed the activity of ALDH, a strong CSC marker. These data are compelling evidence that support the premise that NOTCH1 is a CSC regulator based on demonstrations that in vivo tumor initiation and growth were suppressed when NOTCH1 signaling was blocked by its specific inhibitor, crenigacestat. The results from in vitro and in vivo studies are clear indications that NOTCH1 can modulate CSC activity in ATC.
Comprehensive analyses of available databases of thyroid cancer patients showed that cMYC was expressed at markedly higher levels in ATC than in PTC (Fig. 4A). Of importance, the expression of the cMYC gene and its target genes were positively correlated with CDK7 gene expression (Fig. 4B and Supplementary Fig. S3), suggesting that expression of cMYC would also be sensitive to THZ1 in ATC. Indeed, we found that inhibition of CDK7 and RNAPII phosphorylation by THZ1 resulted in the suppression of cMYC expression at the mRNA and protein level in 11T and 16T cells in vitro as well as within in vivo xenograft tumors. Recent studies reported a selective CDK7 covalent inhibitor, YKL-5-124, that acts to cause cell cycle arrest at the G1/S transition in HAP1 and Jurkat cells, but did not induce any changes in the phosphorylation of RNAPII CTD. 34 –36
We therefore assessed the effects of YKL-5-124 and THZ531, a selective inhibitor of CDK12/1337 on 11T and 16T cells. Of interest, we found that in these two ATC cell lines, YKL-5-124, acted to reduce the phosphorylation of RNAPII CTD, as THZ1 did, but not by THZ531 (Supplementary Fig. S4), indicating the potential off-target effects of THZ1 via CDK12/13 was minimal in ATC cells after treatment with these inhibitors for 24 hours. These findings suggested that the cellular context in different cancer types would provide additional modulation of CDK7-mediated transcription.
As observed in other cancers, 27 –29 we found a functional link of cMYC with NOTCH1 actions in ATC. The gene set enrichment analysis using GO Biological Process (GOBP) showed that the NOTCH1 signaling pathway was highly associated with cMYC expression, which was further demonstrated by the concurrent suppression in the expression of mRNA of both NOTCH1 and cMYC by THZ1 in ATC cells. Moreover, the Stemness Index was positively associated with high expression of NOTCH1 and cMYC in thyroid cancer (TCGA database). Furthermore, we found that the tumorspheres were not only associated with elevated ALDH activity,19 but also with high expression of these two NOTCH1 and cMYC proteins.
Significantly, inhibition of the NOTCH1 signaling by its specific inhibitor, crenigacestat, resulted in the suppression of both cMYC and the active form of NOTCH1, NICD, thereby blocking ATC development by significantly suppressing CSC activity. Together, our studies demonstrated that NOTCH1-cMYC functioned collaboratively to drive ATC progression. Our experiments exemplify how an inhibitor of CDK7 activity can lead to the identification of genes that are critical to ATC progression and sensitive to transcription regulation. The identification of this class of genes (e.g., the NOTCH1 and cMYC genes in this study), when regulated by aberrant transcription, can result in altered functions in CSC activity that drive thyroid cancer progression.
Our proposed model, supported here by our in-depth analysis of TCGA-THCA data, shows that higher CDK7 expression, acting to promoting aberrant transcription through activation of CDK-activating kinase (CAK), is associated with important clinical predictors for thyroid cancer aggressiveness. The advanced symptoms include a higher incidence of extrathyroidal extension (Supplementary Fig. S5A),38 more frequent lymph node metastasis (Supplementary Fig. S5B), 39,40 and ultimately shorter disease-free survival (Fig. 3H). The association was further strengthened by positive correlation in the expression of the two other subunits of CAK, cyclin H encoded by the CCNH gene and CAK assembly factor (MAT1), encoded by the MNAT1 gene (Supplementary Figs. S6 and S7). These observations suggest that targeting transcription via inhibition of CDK7 activity may be a promising strategy for treatment of ATC.
Footnotes
Authors' Contributions
Conception and designs were performed by W.K.L. and S.-Y.C.; Development of methodology and acquisition of data were carried out by W.K.L. and L.Z.; Analysis and interpretation of data were performed by W.K.L. and S-Y.C.; Writing, review, and/or revision were performed by W.K.L. and S.-Y.C. Administrative, technical, or material support were performed by L.Z. and S.-Y.C.
Acknowledgment
The authors thank Joelle Mornini, NIH Library, for article editing assistance.
Data Availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research is supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplemental Information