
Abstract
We report a 10-month-old girl with familial congenital hypothyroidism harboring a novel heterozygous pathogenic variant in the paired DNA-binding domain of PAX8 (NM_003466:c.110T>C:p.Leu37Pro). Genotype–phenotype correlation revealed complete penetrance of this PAX8 defect in this family, in which the affected father and half-brother carry the same mutation. This deleterious variant has not been reported in any of the available databases [MAFgnomAD = 0, dbSNP (−)], and the amino acid leucine at position 37 is highly conserved across species. Establishing the molecular diagnosis expands our knowledge on the cause of thyroid dysgenesis and provides a guide for counseling and early treatment.
Introduction
Primary congenital hypothyroidism (CH) is characterized by elevated levels of thyrotropin (TSH) and reduced levels of thyroid hormones, which, if untreated, lead to intellectual and developmental delay (1). In the majority of cases, the cause is thyroid dysgenesis (TD). Known genetic TD defects are mutations in TSHR, NKX2-1, FOXE1, and PAX8 (1). We report a girl with familial CH caused by an undescribed pathogenic variant in PAX8 found by Sanger sequencing.
Case Report
A 10-month-old girl (Fig. 1A: III-3) of European ancestry living in New Zealand was found to have CH on the second day of life, when serum TSH was >100 mU/L (normal value <20). On the fourth day of life, her TSH level was >300 mU/L, total thyroxine (TT4) and free thyroxine index (fT4I) were low at 5.4 μg/dL (6.1–12.2) and 4.1 (5.8–13.2), respectively. In addition, 99m pertechnetate scan revealed low uptake in two small eutopic lobes (Fig. 1B). Thyroid ultrasonography performed at 17 days of life showed 2 small lobes (right: 5.5 × 3.4 mm and left: 4.9 × 4.5 mm). Ultrasonography of the urinary tract of the proband at the age of one year showed no abnormalities.
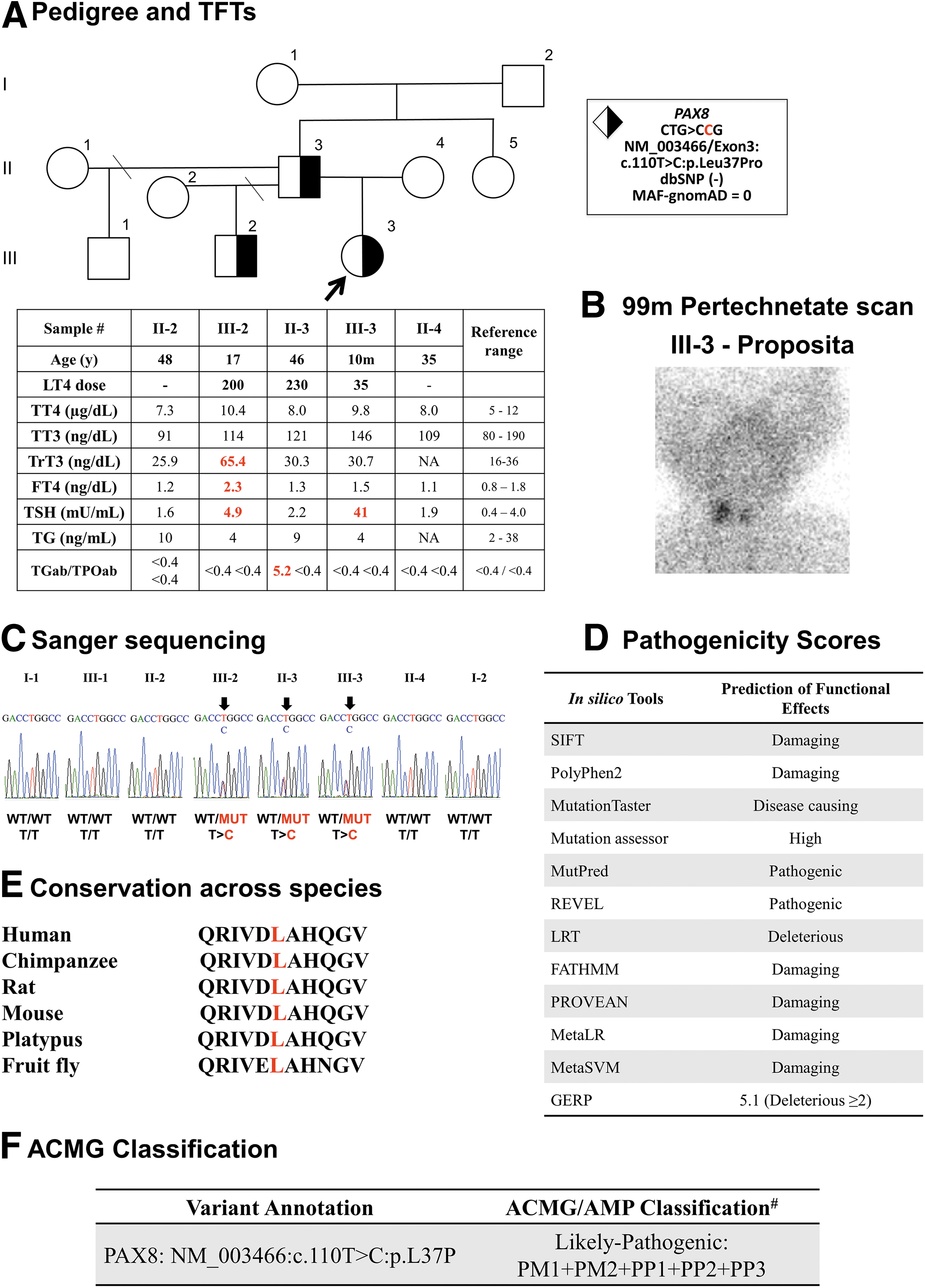
Pedigree of the family with clinical and genetic information. (
Treatment with levothyroxine (LT4) was started with 35 μg/day that was adjusted over time to maintain her thyroid function tests within normal range. Her paternal half-brother (Fig. 1A: III-2) was found to be hypothyroid at 14 days of life when his serum TSH was elevated >100 mU/L, and thyroid hormones were in the lower reference range [TT4 of 6.9 μg/dL (5–19.1) and fT4I of 5.3 (5.8–17.9)]. Radionuclide scan showed two normal eutopic thyroid lobes. At the age of 17 years, he is on 200 μg LT4 daily.
The proposita's father (Fig. 1A: II-3) was found to be hypothyroid at the age of 6 years and was started on LT4 replacement, currently on 230 μg daily average dose. Currently, at the age of 48 years, he shows no obvious intellectual or physical constraints. He completed secondary education and works in real estate. Results of serum thyroid tests obtained at the time of the evaluation are shown in Figure 1A. Familial history revealed that the paternal grandfather (Fig. 1A: I-2) developed autoimmune thyroid disease in later life and has been on LT4 replacement. The paternal grandmother (I-1) has no thyroid abnormalities.
Genetic Analysis
Written informed consents were obtained from all family members before undertaking this investigation and blood sampling. Sanger sequencing of genomic DNA, extracted from circulating leukocytes, was performed using primers flanking all exons of the PAX8 gene. All polymerase chain reaction products were sequenced using BigDye terminator cycle sequencing kit followed by automated sequencing. We identified a heterozygous PAX8 missense variant in PAX8 (ENST00000429538.3/NM_003466/Exon3:c.110T>C:p.Leu37Pro) in the proposita.
Her affected half-brother and father also harbor the variant, whereas all other members of the family, including the paternal grandparents I-1 and I-2, do not carry it. This genotype–phenotype correlation indicates complete penetrance of the PAX8 defect in this family (Fig. 1C). This variant has not been reported in any available database (MAFgnomAD = 0; dbSNP = absent) and it is shown to be deleterious by at least 12 in silico pathogenicity scores (Fig. 1D). In addition, the p.Leu at position 37, located in the paired DNA-binding domain, is highly conserved across species (Fig. 1E). Taken all together, this variant is predicted to be pathogenic according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology classification (2) (Fig. 1F).
Discussion
The first report of loss-of-function PAX8 variants showed 3 heterozygous point mutations located in its paired domain in 2 sporadic patients and 1 familial case of TD (3). Over the years, at least 29 heterozygous mutations in PAX8 have been described in patients presenting predominantly with thyroid hypoplasia (1). The majority of variants are missense located in the paired DNA-binding domain, and inherited among familial cases in an autosomal dominant mode with incomplete penetrance and variable expressivity (1,3). Incomplete penetrance and expressivity could be explained by different mechanisms, such as modifier genes, the influence of the allele in trans, sex, genetic (dominant negative effect or haploinsufficiency), and environmental factors (4).
For instance, seven members of a family harboring the heterozygous PAX8-S48F mutation presented with striking variability in the clinical presentation of CH (5). One of the patients was found to be hypothyroid on neonatal screening and had no thyroidal uptake, whereas her affected brother had normal TSH level upon neonatal screening and was found to have increased TSH value at the age of five years (5). Further evaluation of this mutation showed no loss in DNA binding affinity, but rather a specific defect in transactivation mediated by the coactivator p300. This abnormal cofactor interaction revealed that PAX8-S48F mutant allele competes with PAX8 wild type for DNA binding in the heterozygous state showing that a dominant negative effect could be responsible for the phenotypic expression of the mutated allele (5).
Guided by clinical and genetic analyses, the family herein reported was found to harbor a novel heterozygous missense variant in the paired DNA-binding domain of PAX8, which produced hypoplastic glands leading to congenital nongoitrous hypothyroidism in all affected family members harboring the pathogenic variant. Such dominant pattern of inheritance of full penetrance in familial cases has not been shown by other articles (1,3). Establishing the molecular diagnosis expands our knowledge on the cause of TD and provides counseling regarding early diagnosis and treatment.
Footnotes
Authors' Contributions
A.M.D. and S.R. provided the conceptual framework for the study and designed the experiments. M.M.F. designed and performed the experiments and prepared the first version of the article. L.R. and M.B. identified the patient, gathered the medical information, and recruited members of the family for further studies. All authors reviewed and revised the article.
Acknowledgments
The authors thank the patient and her family for participating in this study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grant DK 15070 from National Institutes of Health, USA, to S.R.