
Abstract
Background:
Nuclear protein in testis (NUT) carcinomas (NC) are a rare, highly aggressive, subset of squamous cell carcinomas, characterized by a translocation involving the NUTM1 gene. Thyroid location of NUT carcinomas has rarely been described.
Methods:
We report here two cases of thyroid NC with NSD3::NUTM1 translocation.
Results:
The first case presented as a very aggressive undifferentiated thyroid carcinoma in a 38-year-old man who died 21 months after the diagnosis. The second case was diagnosed after multiple lymphadenopathy recurrences mainly in the neck in a 37-year-old woman 7 years after total thyroidectomy for papillary thyroid carcinoma with a classic and a solid/trabecular component.
Conclusions:
Our case reports highlight the challenges in diagnosing these exceptional carcinomas. The therapeutic impact of the administration of pharmacological compounds with epigenetic action, in line with the physiopathology of these carcinomas, is also discussed.
Avery rapidly growing thyroid mass with symptoms of mechanical compression is an unusual event, especially in young adults. We report here two cases, both of which proved to be rare malignant diseases, namely NUclear protein in testis (NUT) carcinomas (NC). Our case reports highlight the challenges in diagnosing these exceptional carcinomas and their complex therapeutic management.
Patients
Patient 1
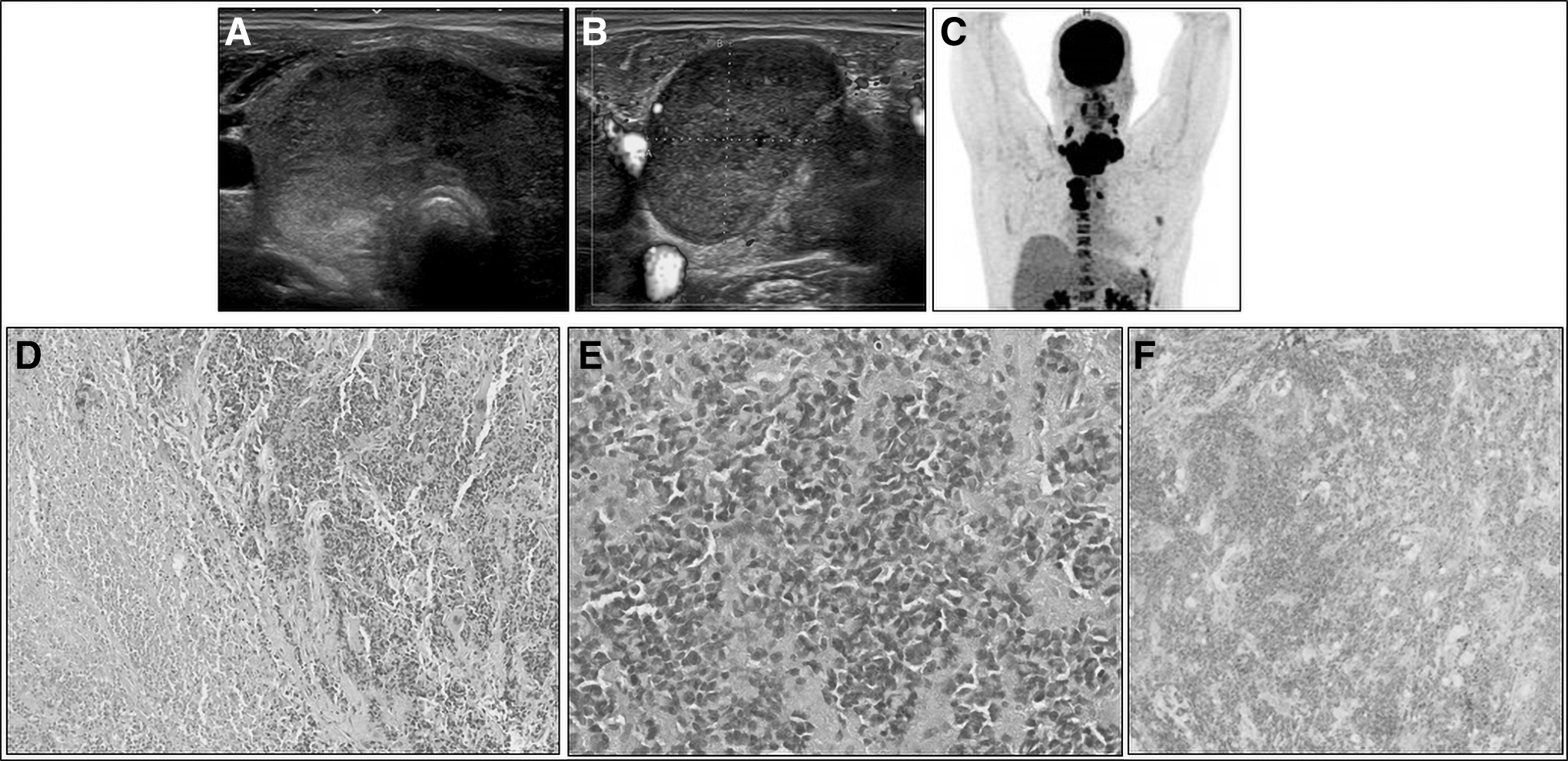
A 38-year-old man was referred for a rapidly progressive cervical mass associated with dysphagia, weight loss, and dysphonia with a left recurrent paralysis. A chest and neck computed tomography (CT) was performed showing a heterogeneous 8.4 cm cervical mass developed at the expense of both thyroid lobes. The mass presented predominantly solid components, with strongly hypoechoic areas on neck ultrasound (Fig. 1A) and was associated with multiple neck (Fig. 1B) and mediastinal lymph nodes measuring up to 3.8 cm. All the lesions had intense fluorodeoxyglucose (FDG) uptake (Fig. 1C).
(
The fine needle aspiration of the tumor mass and of a left IV adenopathy showed the cytological appearance of an undifferentiated carcinoma. As the tumor was totally unresectable, a surgical biopsy was performed. Histological analysis confirmed the undifferentiated features (Fig. 1D, E) infiltrating the striated muscle with tumor necrosis foci. Neither regular thyroid vesicles nor differentiated carcinoma were observed (Fig. 1D). Antigen Ki67 immunostaining showed more than 90% of cells in cycle, supporting the aggressiveness of the tumor.
The cells presented a slightly eosinophilic cytoplasm, giving them a plasmacytoid appearance. Most of the immunostaining were negative: lymphoma markers (CD45, CD3, CD79a, CD20, CD30, CD10, CD138, TdT, and Epstein Barr Virus), germinal tumor marker (SALL4), and differentiated neuro-endocrine tumor markers (Calcitonin, chromogranin, and synaptophysin). Immunohistochemistry revealed cytokeratin AE1/AE3 heterogeneous positivity, TTF1 moderate positivity, and PAX8 marking rare nuclei, raising the hypothesis of an atypical anaplastic thyroid carcinoma. However, CD99 positive immunostaining led to the hypothesis of a tumor belonging to the Ewing sarcoma family (ESFT).
Next generation sequencing (NGS) was performed on formalin-fixed paraffin-embedded (FFPE) tissue searching for DNA mutations in 41 genes and for 284 different fusion genes (Supplementary Data S1). The only identified molecular alteration was a variant of unknown significance in the ALK gene (ALK 3692G>A). After discussion in a multidisciplinary staff meeting, the patient qualified for neoadjuvant chemotherapy according to the Euro Ewing 2012 protocol 1 usually used for ESFT.
After the first cycle, although the clinical symptoms had already improved the complementary molecular testing, that is, RNA sequencing revealed a fusion transcript NSD3::NUTM1, leading to the final diagnosis of an NC of thyroid origin. Immunostaining with an anti-NUT antibody on the biopsy tissue showed positivity (Fig. 1F). Due to the primary satisfying clinical response, the same chemotherapy regimen was continued to be associated with conventionally fractionated intensity modulated radiation therapy.
An almost complete morpho-metabolic response was obtained after the sixth cycle. One month after the seventh cycle, a new FDG uptake of suspicious left level IV lymph nodes was observed. There was also tumor infiltration of almost all the left lobe without FDG uptake, which was confirmed on neck ultrasound and CT. The patient was referred for surgery limited to a left lobectomy because of left recurrent nerve hypomobility and the absence of suspicious contralateral lesions. Histological analysis of the tissue showed no residual lesion, and NUT antibody negativity.
Centimeter locoregional recurrence and mediastinal lymph nodes as well as a new non-hypermetabolic 6 mm-lung nodule were detected three months postoperatively on FDG-positron emission tomography (FDG-PET).
The patient was included in a phase I/II therapeutic trial testing a bromo-domain inhibitor molibresib (NCT01587703). 2 Within two months, after two cycles of treatment, a symptomatic decrease in left ventricular function down to 43% with dyspnea led to the bromodomain inhibitor withdrawal and exclusion of the patient from the protocol. Left ventricular function improved to 50%, 15 days after the treatment withdrawal. After diffuse progression of the disease (left thyroid bed, mediastinal adenopathys, pleural, and pulmonary metastases), he was treated with carboplatin/taxol chemotherapy for two cycles but died 21 months after initial diagnosis.
Patient 2
A 37-year-old woman was diagnosed with a 47 mm pT3aN1a papillary thyroid carcinoma (PTC) with a classic component and a solid/trabecular component. However, the evaluation of the mitotic index, as recommended by the WHO 2022 Classification of Thyroid Neoplasms, 3 was not possible. She was treated by a total thyroidectomy with bilateral central compartment neck-dissection. The surgery was followed by a 3.7 GBq adjuvant radioiodine therapy.
Ultrasound was normal, and the post-therapeutic whole-body scan (WBS) showed a central neck uptake. Ultrasensitive thyroglobulin (Tgus) concentration was 0.4 μg/L, without Tg antibodies (thyrotropin [TSH]: 140 mUI/L). The patient was treated with neck re-operations two and four years after the initial diagnosis, revealing multiple PTC lymph node metastases measuring up to 27 mm. A second 3.7 GBq radioiodine treatment was performed after the second surgery, revealing no uptake on the WBS with undetectable Tgus.
Two years after the third surgery, neck ultrasound, neck and chest-CT and FDG-PET revealed multiple, highly suspicious lymph nodes in the IV and V inferior neck levels measuring up to 22 mm with axillary lymph nodes (Fig. 2A), all presenting FDG uptake (Fig. 2B). Tgus concentration was 19 μg/L (TSH 0.01 mUI/L, without Tg antibodies). The NGS performed on the FFPE tissue of one of the lymph nodes searching for DNA mutations in 50 genes revealed only a c.262G>A CDKN2A mutation (Supplementary Data S2).

(
Fusion transcripts in 14 genes were also searched for by NGS (Supplementary Data S2) on the FFPE tissue from a 20 mm right level IV lymphadenopathy surgically resected for that purpose. An expert pathologist diagnosed poorly differentiated cancer in the lymph node. A few months later, a fourth operation consisting of right level IV neck dissection with right axillary dissection was performed. Histology revealed 11 poorly differentiated metastatic lymph nodes (Fig. 2C) measuring up to 5 cm with diffuse TTF1 (Fig. 2D), CD99 and p53 positivity.
Immunohistochemistry with p40, p63 was positive (15% and 25% of the tumoral cells, respectively) and CD34 was negative. Antigen Ki67 immunostaining showed 10% of cells in cycle. Postoperative Tgus was 1.5 μg/L (TSH: 0.1 mUI/L). Exome analysis and RNA Sequencing after inclusion in the “EXORARE” program (Supplementary Data S3) was performed revealing NSD3::NUTM1 translocation, leading to the diagnosis of metastatic NC.
Pathogenic mutations in CDKN2A, SMARCA4, NOTCH2, KDM6A, and SPOP genes were also identified. Immunostaining with an anti-NUT antibody was then performed and was negative despite the use of two different techniques (Fig. 2E). Neck and chest-CT performed one year after the fourth surgery revealed multiple suspicious right supraclavicular, subclavian, and axillary lymph nodes measuring up to 27 mm remaining relatively stable on neck ultrasound and CT performed six months later. There were no other lesions. Tg concentration was 15 μg/L (TSH: 0.01 mUI/L). At the last visit, the patient was still not receiving systemic treatment.
Discussion
The NC, which was first described in 1991, 4 is a very rare, aggressive poorly differentiated to undifferentiated carcinoma. The incidence is higher in children and young adults (2), with both sexes equally affected. 5 Hematogenous distant metastases are frequent, and NC are usually rapidly lethal. The median overall survival (OS) is 5–6.7 months in the largest cohorts (119 and 54 patients, respectively). 5,6
The cellular origin is a cause for debate, and NC might derive from an epithelial cell or is also recognized as a subtype of squamous cell carcinoma. 7 The NC was previously called NUT midline carcinoma because it occurred mainly in the midline structures above the diaphragm such as head, neck, and mediastinum. 8,9 However, various locations have been reported such as the pancreas, kidney, and bladder. 10,11 The thyroid location is uncommon and has been rarely described. 12 –16 For patients reported here, our hypothesis is that NUT fusion occurred in a thyrocyte driving the development of aggressive carcinomas.
The moderate TTF1 and PAX8 positivity in the first case and the detectable Tgus levels and TTF1 positive Immunohistochemistry in the second case support our hypothesis. Unfortunately, the search for NUT fusion in the primary tumor was not possible for case 2. However the tissue of the primary tumor shares histological features with the metastatic lymph nodes in which the NSD3::NUTM1 fusion was identified. Among the previously reported cases, 12 –16 the hypothesis of an NUT fusion occurring in a thyrocyte may be considered in three of them. 12,15,16
The NC is genetically defined by a chromosomal rearrangement involving the NUTM1 gene (NUT midline carcinoma family member 1) coding for a protein that physiologically binds to histone acetyltransferase p300, resulting in histone acetylation. NUTM1 translocation occurs mostly with genes encoding for NH2 terminal bromodomain and extra terminal domain protein (BET proteins). 17 In 70% of NC, Bromodomain-containing protein 4 gene (BRD4) is involved. 18
In other cases, fusions are called NUT-variant proteins, such as in our cases, with NUTM1 translocation involving NSD3, encoding for NSD3 Histone-Lysine N-Methyltransferase. BRD4::NUT fusion proteins act to maintain growth and block cell differentiation by interacting with chromatin, leading to deregulation of different genes expression and in particular MYC. 18 Interestingly, NSD3 function depends on BRD4 and its interaction with chromatin. 19
The absence of multiple significant molecular alteration in our patients, also reported by Allison et al, 12 contrasts with other undifferentiated carcinomas that accumulate genetic alterations, so-called late events, especially TP53 and TERT promoter mutations. Classical NUT midline carcinomas from the head and neck harbor mostly a single driving NUTM1 fusion with a few, if any, additional oncogenic mutations, suggesting that NUT fusion is an early event that is alone sufficient to trigger the carcinogenesis. 18,20
The NC diagnosis is challenging. Conventional and functional imaging (i.e., FDG-PET) are nonspecific. 21 Regarding histology, specific morphological features are lacking, with frequent delay in NC diagnosis, as in our cases. Histopathological aspects usually consist of monomorphic undifferentiated small- to medium-sized round or oval cells with a high nuclear-to-cytoplasm ratio. The rather clonal and uniform appearance and the focal abrupt keratinization with expression of cytokeratin proteins are significant pathological characteristics of NC 18,22,23 but not specific enough to allow a definitive diagnosis.
The NC can be diagnosed by immunohistochemistry or fluorescent in situ hybridization (FISH) with a specific anti-NUT protein antibody, currently commercially available (NUT[C52B1] monoclonal Rabbit antibody; Cell Signaling Technologies #3625) with a sensitivity and specificity of 87% and 100%, respectively. 24 The 87% sensitivity of this Immunohistochemistry technique probably explains the negative result for the second case, as already reported earlier, 12 suggesting that RNA sequencing should be systematically performed in similar situations. Immunostainings with p40, p63, and CD99 for case 2 were positive, as already described in NUT carcinoma.
The diagnostic challenge is to determine when to perform Immunohistochemistry, FISH, or molecular tests leading to the identification of NUT fusions. The NC may be underestimated, and to raise awareness and disseminate information, an international NUT midline carcinoma registry (
To date, no therapeutic strategy is approved for NC. The current adopted practice is to perform a multimodal approach with chemotherapy, surgery, and radiation therapy that may improve progression-free survival and OS. 5,25 A few cases have been treated as sarcoma, similarly to our first case 26 with complete remission in two cases for more than 6 27 and 13 years, respectively. 28 New targeted therapies have emerged, such as histone deacetylase inhibitors and BET inhibitors; some of them are investigated in phase I/II clinical trials. 2,29 –31
The follow-up of only three young patients, between 34- and 42-years old, with thyroid NC has been reported so far. One of them was in complete remission 18 months after the diagnosis followed by neck radio-chemotherapy and treatment with molibresib, a BET inhibitor. 13 The third patient died 10 months after the diagnosis followed by chemotherapy and the anti-PD1 camrelizumab. 16 The development of NSD3 inhibitors may represent an interesting therapeutic approach. 32
In conclusion, thyroid NC is a challenging diagnosis, made possible preferentially by RNA sequencing. Clinicians and pathologists should be aware of this rare tumor, possibly underdiagnosed among undifferentiated thyroid tumors without classical molecular alterations identified on large NGS panels. Despite too few cases to establish optimal therapeutic management, the diagnosis may enable the patient's inclusion in early phase trials of pharmacological compounds targeting epigenetic pathways. Thus, the search for NUT fusions by routine NGS panels should be considered.
Footnotes
Authors' Contributions
N.S.: investigation, visualization, and writing—original draft; J.W.: resources, writing—review and editing; C.T.: resources, writing—review and editing; E.G.: resources, writing—review and editing; G.D.: resources, visualization, and writing—review and editing; B.C.-P.: resources, visualization, and writing—review and editing; N.C.: resources, writing-review, and editing; S.G.: resources, writing—review and editing; J.-M.S.: writing—review and editing; L.L.: conceptualization, writing—review and editing; L.G.: conceptualization, project administration, resources, and writing—review and editing; and C.B.: conceptualization, supervision, visualization, project administration, resources, and writing—review and editing.
Acknowledgment
The authors thank Liz Atzel for editing the English version of this article.
Author Disclosure Statement
No competing financial interests exist.
Statement of Ethics
Consent has been obtained from each patient after full explanation of the purpose and nature of all procedures used (Ethics Committee Approval CLEP AAA-2022-08032).
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Data S1
Supplementary Data S2
Supplementary Data S3