
Abstract
Background:
Metastatic disease is a major cause of thyroid cancer-related death. However, the mechanisms responsible for thyroid cancer metastasis are unclear. Dipeptidyl peptidase-4 (DPP4) is a multifunctional cell surface glycoprotein that has been reported to be a negative prognostic factor in thyroid cancer. We explored the molecular mechanism of the role of DPP4 in thyroid cancer cell metastasis.
Methods:
The effects of DPP4 on thyroid cancer cell migration/invasion in vitro were assessed by transwell assays. A lung metastatic mouse model was also established to determine the effect of DPP4 on tumor metastasis in vivo. DPP4 inhibitor sitagliptin was used to test its effect on thyroid cancer cell metastasis. The mechanism of which DPP4 promotes thyroid cancer cell metastasis was explored by a series of molecular and biochemical experiments.
Results:
We observed that DPP4 was significantly upregulated in papillary thyroid cancers compared with control subjects, and its expression was positively associated with lymph node metastasis and BRAFV600E mutation. Functional studies showed that DPP4 knockdown significantly inhibited metastatic potential of thyroid cancer cells, and vice versa. However, DPP4 inhibitor sitagliptin did not affect the metastatic ability of thyroid cancer cells, indicating that the promoting effect of DPP4 on tumor metastasis was independent of its enzymatic activity. Mechanistically, DPP4 interacted with the α4 and β1 integrin subunits, and stabilized the formation of integrin α4β1 complex. DPP4-mediated integrin signal activation promoted the nuclear localization of c-Jun through the FAK/AKT pathway, thereby inducing the transcription of transforming growth factor-beta 1 (TGFB1, coding for protein TGF-β1). TGF-β1 then facilitated tumor metastasis by inducing the epithelial–mesenchymal transition.
Conclusions:
DPP4 promotes thyroid cancer cell metastasis through the integrins/FAK/AKT/c-Jun/TGF-β1 signaling axis. These findings may have implications for an alternative therapeutic strategy for thyroid cancer.
Introduction
Thyroid cancer-related death is mainly caused by tumor metastasis rather than the primary tumor itself. 1,2 A report analyzing the SEER database showed that the five-year survival rates of thyroid cancer were 99.9% for localized tumors, 98.3% for regional staged tumors, but dropped down to 53.3% for distantly metastatic tumors. 3 Given the high death risk of metastatic thyroid cancers, a better understanding of the mechanisms involved in thyroid cancer metastasis is important.
Aberrant activation of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway by some genetic alterations plays a pivotal role in thyroid tumorigenesis and progression. 4 Among them, BRAFV600E mutation is one of the most important molecular events that cause the overactivation of the MAPK/ERK pathway. 5 A large number of studies demonstrated that BRAF mutations increased the aggressive behaviors of thyroid cancer, including extrathyroidal extension and lymph node and distant metastasis. 6 –9 Besides, BRAF mutations induced different gene expression profiles, thereby promoting tumor progression by altering biological activities of cancer cells. 7
Dipeptidyl peptidase-4 (DPP4; also known as CD26) is a member of the prolyl oligopeptidase family, which can cleave N-terminal dipeptides with either penultimate proline or alanine. 10 As a multifunctional cell surface type-II membrane glycoprotein, DPP4 can also interact with other molecules such as seprase, ADA, and CD45 in an enzymatic activity-independent manner. 11 –13 The diversity of its substrates, together with its nonenzymatic functions, explains its involvement in various physical and pathological processes such as glucose and lipid metabolism, immune modulation, and tumorigenesis. 12,14,15 However, the role of DPP4 in thyroid cancer cell metastasis has not been defined.
Materials and Methods
Clinical specimens
This study obtained approval from the Institutional Review Board and Human Ethics Committee of the First Affiliated Hospital of Xi'an Jiaotong University (No. 2017-43), and informed consent was obtained from all participants for tissue collection. A total of 20 pairs of primary papillary thyroid cancer (PTC) and adjacent noncancerous tissues were obtained from patients from the First Affiliated Hospital of Xi'an Jiaotong University. None of the participants had received any systemic therapy before surgery.
Measurement of DPP4 enzymatic activity
BCPAP and 8305C cells (1 × 106) were suspended in phosphate-buffered saline (PBS) and seeded in 96-well plates. Cells were then treated with or without 20 μM of DPP4 inhibitor sitagliptin for 2 hours after which Gly-Pro p-nitroanilide hydrochloride (at 0.5 mM, G0513; Sigma-Aldrich) was added as a chromogenic substrate and the reaction was incubated at 37°C for 2 hours. The optical density values were monitored at 405 nm using a spectrophotometer.
Animal studies
All mouse experiments were conducted by following the NIH guidelines and approved by the Ethics Committee of Biological and Medical Study of Xi'an Jiaotong University (No. 2017-442). Appropriate measures were taken to ensure minimal pain or discomfort in animals. The article was prepared in accordance with the ARRIVE Guidelines 2.0.
To establish a lung metastatic mouse model, seven-week-old female
Nuclear and cytoplasmic protein extraction
Nuclear and cytoplasmic extracts were prepared using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Shanghai, China). Briefly, 2 × 106 cells were washed with PBS and resuspended in 200 μL of cytoplasmic protein isolation solution A supplemented with phenylmethylsulfonyl fluoride and homogenized by vortexing. After 15 minutes on ice, 10 μL of cytoplasmic protein isolation solution B was added, and cells were then homogenized on ice. Next, the complex was centrifuged at 12,000 g for 5 minutes at 4°C. The resulting supernatant was a cytoplasmic protein fraction. The pellet was then resuspended in 50 μL of nuclear protein isolation solution supplemented with phenylmethylsulfonyl fluoride, homogenized on ice using an ultrasonic crusher, and centrifuged at 12,000 g for 10 minutes at 4°C. The resulting supernatant was a nuclear protein fraction.
Chromatin immunoprecipitation–quantitative polymerase chain reaction
The detailed protocols were conducted as described previously. 16 The primer sequences for quantitative polymerase chain reaction (qPCR) are summarized in Supplementary Table S1.
Coimmunoprecipitation
The indicated antibodies or IgG was added into equal amount of lysates, and then incubated at 4°C overnight. On the second day, protein A/G plus-agarose beads (Catalog#: sc-2003; Santa Cruz, CA) were added into the above lysates and then incubated at 4°C for 5 hours. Next, the samples were washed five times and then subjected to Western blot analysis. All of the antibodies are presented in Supplementary Table S2. Additional methods are presented in Supplementary Data.
Results
Increased expression of DPP4 is related to lymph node metastasis in thyroid cancer patients
We first analyzed mRNA expression of DPP4 in 20 pairs of primary PTCs and their matched noncancerous tissues, and found that its expression was significantly increased in the former (p = 0.009) (Fig. 1A). Next, we selected 11 pairs of samples and investigated protein expression of DPP4 by immunohistochemistry (IHC) assays. As expected, its protein expression was also elevated in PTCs compared with their matched noncancerous tissues (Fig. 1B). These results were in accordance with the data from The Cancer Genome Atlas (TCGA) database and Gene Expression Omnibus (GEO) database showing that DPP4 expression was markedly increased in PTCs compared with control subjects (Fig. 1C and Supplementary Fig. S1A). Data from the GEO database also showed increased expression of DPP4 in follicular thyroid cancers and anaplastic thyroid cancers (Supplementary Fig. S1B, C).

Increased expression of DPP4 in PTCs. (
Next, we analyzed DPP4 expression in different subtypes of PTC using the TCGA database, including conventional PTCs, follicular variant PTCs, and tall-cell PTCs (TCPTCs). We found that its expression in TCPTCs was the highest compared with control subjects or other subtypes (Fig. 1D). Therefore, we hypothesized that DPP4 might be related to the aggressive phenotypes of thyroid cancer cells. We further analyzed data from the TCGA database and found that DPP4 expression was higher in PTC patients with lymph node metastasis than those without lymph node metastasis (Fig. 1E), suggesting that there may be a relationship between increased DPP4 expression and thyroid cancer cell metastasis.
DPP4 is transcriptionally regulated by BRAFV600E-induced activation of MAPK/ERK pathway
BRAF or RAS mutations are two major genetic events in thyroid tumorigenesis and progression, and strongly related to malignant phenotypes of thyroid cancer cells including lymph node metastasis and distant metastasis. 17,18 We thus evaluated the association of DPP4 expression with these two molecular events using the TCGA database. The results showed that DPP4 expression was abnormally elevated in BRAF V600E -mutated PTCs compared with RAS-mutated or wild-type PTCs (Fig. 2A), suggesting that DPP4 may be transcriptionally regulated by BRAF mutation in thyroid cancer. To prove this, we ectopically expressed wild-type and V600E mutant BRAF in 293T cells. The results showed that, compared with empty vector-transfected 293T cells, DPP4 expression was significantly elevated in BRAFV600E-expressing cells, but not in cells expressing wild-type BRAF (Fig. 2B).

Upregulation of DPP4 by BRAFV600E-driven MAPK/ERK cascade. (
In line with the above results, we analyzed Dpp4 expression in tumor tissues from transgenic mice specifically expressing BRAFV600E and control mice by IHC assays (n = 5/group), and demonstrated increased expression of Dpp4 in the former (Fig. 2C).
We treated BRAFV600E-mutated BCPAP and 8305C cells with 1 μM PLX4720, a selective BRAFV600E inhibitor, in combination with 500 nM GSK112021, an MEK1/2 inhibitor, to completely block the MAPK/ERK signaling pathway. The results showed that the blockade of MAPK/ERK pathway dramatically decreased DPP4 expression at both mRNA and protein levels (Fig. 2D, E), further supporting the above conclusions.
DPP4 promotes thyroid cancer cell metastasis in vitro and in vivo
We next evaluated the effect of DPP4 on thyroid cancer cell migration and invasion. First, we knocked down DPP4 by two specific siRNAs (si-DPP4 #1 and si-DPP4 #2) in BCPAP and 8305C cells; and in the meanwhile, ectopically expressed DPP4 in IHH4 cells (Fig. 3A, B). The results showed that DPP4 knockdown suppressed thyroid cancer cell migration/invasion ability compared with the control (Fig. 3C). Conversely, ectopic expression of DPP4 enhanced thyroid cancer cell invasiveness compared with the control (Fig. 3D). We treated BCPAP and 8305C cell with DPP4 inhibitor sitagliptin, and demonstrated that sitagliptin significantly inhibited DPP4 activity compared with the control (Supplementary Fig. S2). However, we observed no significant effect of sitagliptin on cell migration and invasion (Fig. 3E, F). These results suggest that the promoting effect of DPP4 on thyroid cancer cell invasiveness is independent of its enzymatic activity.
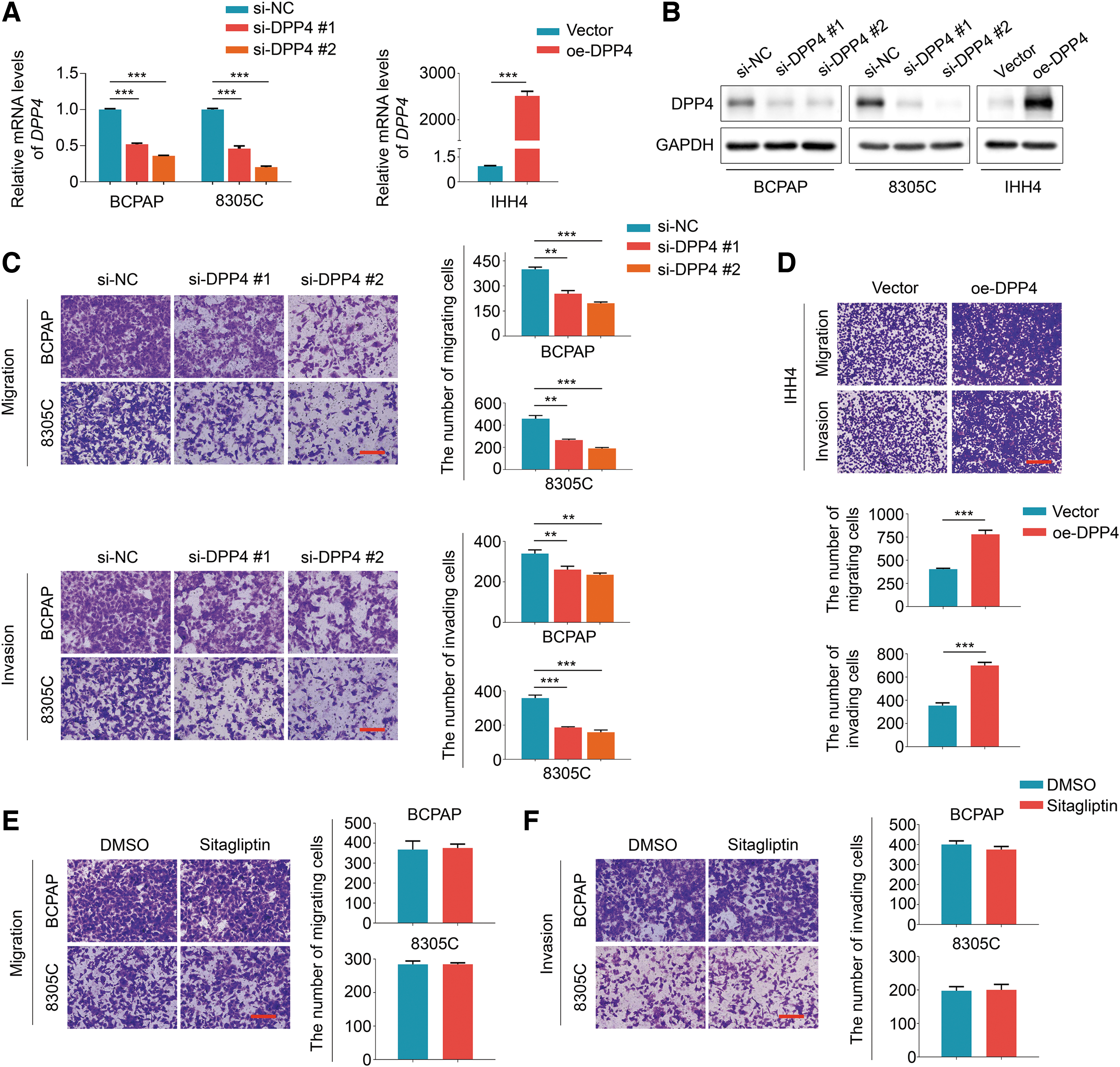
DPP4 enhances thyroid cancer cell migration and invasion in vitro. qRT-PCR (
We established a lung metastatic mouse model by injecting luciferase-expressing DPP4-knockdown 8305C cells and control cells into NPG mice through the tail vein, and evaluated the effect of DPP4 knockdown on tumor metastasis. The results showed that the luminescence intensity in lung of mice bearing DPP4-knockdown tumors was significantly weaker than that of control mice on the 30th day after injection (Fig. 4A). Moreover, we observed bone metastasis in one of the control mice, but none in mice bearing DPP4-knockdown tumors (Fig. 4A and Supplementary Fig. S3).

DPP4 promotes thyroid cancer cell metastasis in vivo. (
At the end of the experiments, the lungs of mice were rapidly harvested and tested by bioluminescence imaging. The results showed that the DPP4-knockdown group had fewer lung metastatic foci and a lower luminescence signal than the control group (Fig. 4B). Moreover, lung weight was significantly lower in the DPP4-knockdown group compared with the control group (Fig. 4C), while body weight did not significantly differ between groups (Fig. 4D).
We also performed hematoxylin and eosin staining in tumor tissues from DPP4-knockdown group and control group, and found that the number and size of lung metastatic foci were significantly decreased upon DPP4 knockdown, and DPP4-knockdown group exhibited relatively normal lungs without obvious tissue damage and remodeling (Fig. 4E). The staining of exogenous luciferase was used to distinguish tumor nodules from lung tissues. We found that DPP4 expression was clearly decreased in metastatic foci from DPP4-knockdown group compared with control group (Fig. 4E). These results, taken together, further support the metastasis-promoting effect of DPP4 in thyroid cancer.
DPP4 induces transforming growth factor-beta 1 expression and epithelial–mesenchymal transition through the AKT/c-Jun signaling pathway
Given that epithelial–mesenchymal transition (EMT) is a key process for cancer cell metastasis, 19 we thus studied the effect of DPP4 on the expression of EMT-related molecules in thyroid cancer cells. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assays showed that DPP4 knockdown significantly decreased the expression of N-Cadherin and Vimentin, while elevated the expression of E-Cadherin in BCPAP and 8305C cells, and vice versa (Supplementary Fig. S4A). This was also supported by Western blotting (Fig. 5A). Meanwhile, we also assessed the effect of DPP4 on the expression of EMT-related transcription factors including TWIST, SNAIL, and SLUG and matrix metalloproteinases (MMPs) including MMP2 and MMP9. The results showed that DPP4 knockdown strongly inhibited the expression of these molecules compared with the control, and vice versa (Supplementary Fig. S4B, C).
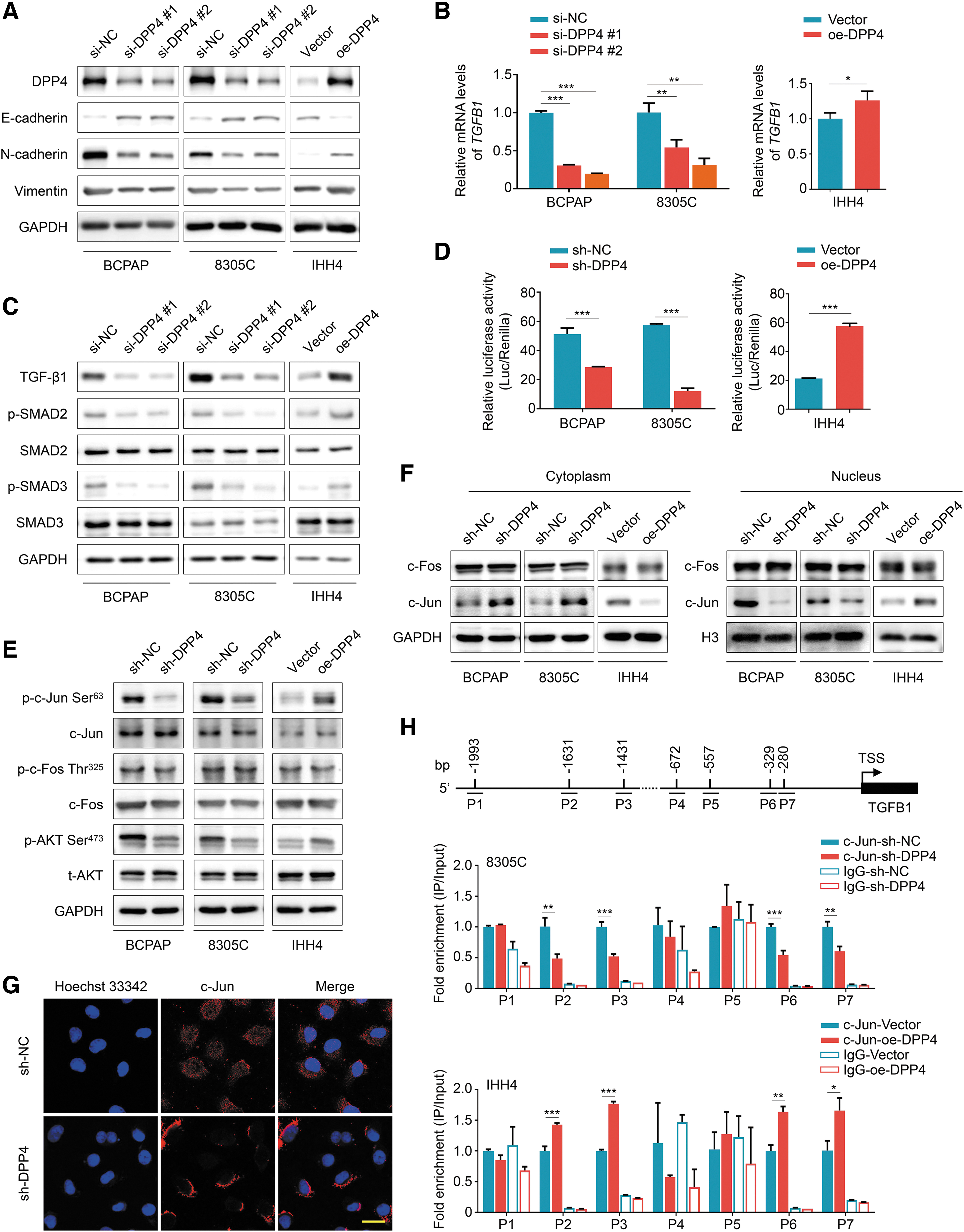
DPP4-mediated TGF-β1 upregulation and EMT through the AKT/c-Jun signaling pathway. (
Transforming growth factor-beta (TGF-β) has been demonstrated to be a crucial regulator of EMT 20 ; we next assessed the effect of DPP4 on its expression in thyroid cancer cells. The results showed that DPP4 knockdown decreased TGF-β1 expression and suppressed its downstream TGF-β1/SMAD ignaling pathway, and vice versa (Fig. 5B, C), suggesting that TGFB1 (coding for protein TGF-β1) may be transcriptionally regulated by DPP4. To determine how DPP4 regulates TGFB1 transcription, we explored the regulatory elements within TGFB1 promoter using the Eukaryotic Promoter Database (EPD). We found that its promoter contained 15 putative binding sites of activating protein-1 (AP-1). These observations suggest that DPP4 promotes TGFB1 transcription by modulating AP-1 activity. To prove this, we first developed a luciferase reporter plasmid containing three canonical AP-1 binding sites, and then assessed the effect of DPP4 on the transcriptional activity of AP-1 in thyroid cancer cells.
The results showed that DPP4 knockdown significantly repressed the transcriptional activity of AP-1 compared with the control, and vice versa (Fig. 5D), further supporting the above conclusion.
AP-1 proteins are homodimers and heterodimers that comprised basic region-leucine zipper proteins. 21 The main AP-1 proteins are c-Jun and c-Fos. 22 To explore the mechanism by which DPP4 modulates AP-1 activity, we evaluated the effect of DPP4 on the expression and phosphorylation of these two components of AP-1. As shown in Figure 5E, DPP4 knockdown or overexpression did not change their expression and c-Fos phosphorylation; however, DPP4 knockdown strongly repressed c-Jun phosphorylation compared with the control, and vice versa.
We found that DPP4 knockdown inhibited the phosphorylation of AKT at Ser473, while DPP4 overexpression elevated its phosphorylation compared with the control (Fig. 5E). These results were also supported by the IHC results in lung metastatic foci (Supplementary Fig. S5). We also observed that DPP4 knockdown decreased the nuclear protein levels of c-Jun and increased its cytosolic protein levels, while it did not change c-Fos distribution between the nucleus and cytoplasm, and vice versa (Fig. 5F). These findings were supported by the results of immunofluorescence assays (Fig. 5G).
To determine direct regulation of TGFB1 by c-Jun through binding to its promoter, we performed chromatin immunoprecipitation (ChIP) assays using the c-Jun antibody, followed by qPCR targeting the seven regions within its promoter predicted by EPD (Fig. 5H, upper panel). The results showed that DPP4 knockdown in 8305C cells significantly reduced the binding of c-Jun to the four fragments (P2: −1664/−1563; P3: −1432/−1361; P6: −420/−302; and P7: −321/−236) within the TGFB1 promoter, while it did not affect its enrichment to the other three fragments (P1: −1664/−1563; P4: −1432/−1361; and P5: −321/−236) (Fig. 5H, middle panel). On the contrary, the recruitment of c-Jun to TGFB1 promoter was expectedly elevated in DPP4-overexpressed IHH4 cells compared with the control cells (Fig. 5H, lower panel), further supporting TGFB1 as a direct target of c-Jun. Collectively, the above results indicate that DPP4 promotes TGF-β1 expression and the EMT process through the AKT/c-Jun signaling pathway.
DPP4 activates the FAK/AKT signaling by interacting with and stabilizing integrin α4β1 complex
We hypothesized that DPP4 interacts with integrin α4β1 complex to activate the FAK/AKT signaling, thereby promoting thyroid cancer cell metastasis. We performed a series of coimmunoprecipitation assays and demonstrated that there were interactions among DPP4, integrin α4, and integrin β1 (Fig. 6A–C). Further studies found that DPP4 knockdown attenuated the interaction between integrin β1 and α4 subunits, while DPP4 overexpression enhanced the formation of integrin α4β1 complex (Fig. 6D). However, DPP4 inhibitor sitagliptin did not affect the interaction between integrin α4 and β1 (Supplementary Fig. S6). These results imply that DPP4 interacts with and stabilizes the integrin α4β1 complex, and this effect is independent of its enzymatic activity.
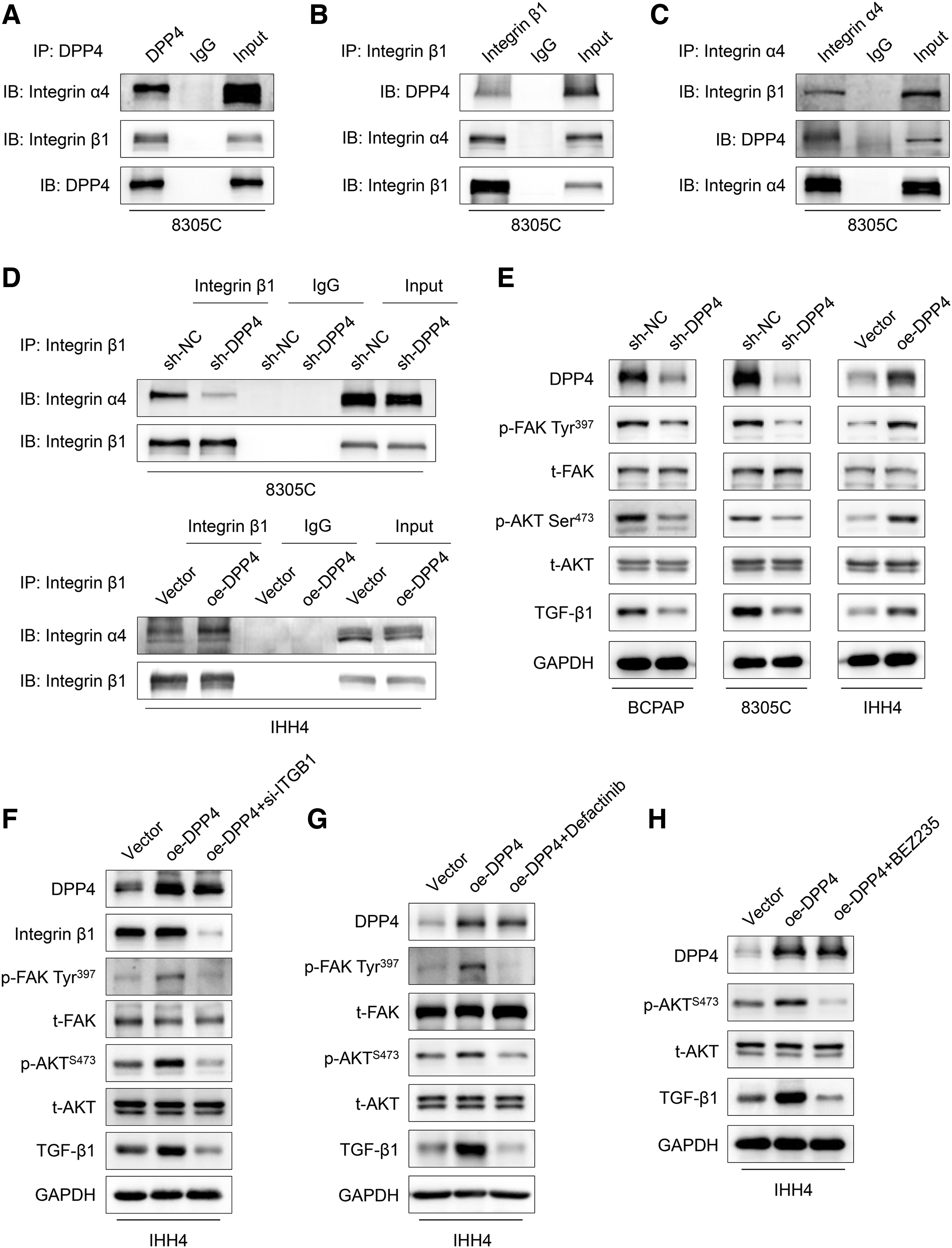
DPP4 stabilizes the integrin α4β1 complex and activates its downstream signaling pathway. (
We next investigated the effect of DPP4 on the activity of FAK signaling pathway. The results showed that DPP4 knockdown repressed the phosphorylation of FAK at Tyr397 and decreased AKT phosphorylation and TGF-β1 expression compared with the control, and vice versa (Fig. 6E). To further prove these conclusions, we knocked down ITGB1 coding for integrin β1 in DPP4-overexpression IHH4 cells, and found that DPP4 overexpression upregulated the phosphorylation of FAK and AKT and the expression of TGF-β1, while ITGB1 knockdown attenuated this effect (Fig. 6F). Next, we treated DPP4-overexpression IHH4 cells with an FAK inhibitor defactinib, and found that defactinib effectively suppressed FAK phosphorylation and reversed the promoting effect of DPP4 overexpression on AKT phosphorylation and TGF-β1 expression compared with the control (Fig. 6G). In addition, we also treated the above cells with BEZ235, an inhibitor of PI3K/mTOR, and found that BEZ235 significantly blocked AKT activation, and, in the meanwhile, reversed DPP4 overexpression-induced TGF-β1 upregulation in comparison with control (Fig. 6H).
Discussion
We found that DPP4 expression was elevated in PTCs compared with control subjects, and positively associated with the presence of BRAFV600E mutation. Previous studies also showed that DPP4 was highly expressed in thyroid cancers. 23,24 We also observed that DPP4 was transcriptionally regulated by BRAFV600E-driven activation of MAPK/ERK pathway. A previous study showed that DPP4 was downregulated by adenosine in the colon cancer cell line HT-29 through the blockade of ERK1/2 signaling. 25 We found a strong association between DPP4 expression and lymph node metastasis by analyzing the TCGA database. This was supported by our data demonstrating that DPP4 enhanced thyroid cancer cell metastasis in vitro and in vivo.
In recent years, there are studies showing that DPP4 inhibitor sitagliptin enhanced tumor immunity by preserving functional chemokines CXCL10 and IL-33. 26,27 Other studies indicated that DPP4 inhibitors could increase the metastatic potential of breast cancer, liver cancer, and colon cancer. 28,29 These observations may explain DPP4 having many substrates. 23 We determined the potential role of DPP4 inhibitor in thyroid cancer cell metastasis. Unlike previous studies, 28,29 our data showed that DPP4 inhibitor sitagliptin had little effect on thyroid cancer cell metastasis, indicating that the promoting effect of DPP4 on thyroid cancer cell metastasis is independent of its enzymatic activity.
EMT is considered to be a crucial process for the dissemination of cancer cells, and closely related to metastasis in epithelial tumors. 30,31 We demonstrated that DPP4 induced EMT in thyroid cancer cells, which was consistent with a previous study. 32 It is well-documented that TGF-β is a potent EMT inducer. 20 We showed that DPP4 indeed transcriptionally upregulated TGF-β1. In analyzing the sequence of TGFB1 promoter, we found 15 canonical AP-1 binding sites in its proximal promoter, and as supported by previous studies, TGFB1 could be directly regulated by AP-1. 33,34 We also showed that DPP4 increased transcriptional activity of AP-1, but did not change the expression of its two subunits c-Jun and c-Fos.
Furthermore, our results demonstrated that DPP4 increased the phosphorylation of c-Jun and promoted its nuclear localization. Our data showed that DPP4 increased the phosphorylation of AKT, which is consistent with a prior report indicating that activated AKT phosphorylates c-Jun and promotes its nuclear localization. 35 We performed ChIP assays to determine direct regulation of TGFB1 by c-Jun. Although seven putative binding sites of c-Jun were predicted in TGFB1 promoter by EPD, our results demonstrated that four of them were responsible for TGFB1 transcription. These observations suggest that DPP4 increases TGF-β1 expression and subsequently promotes the EMT process through the AKT/c-Jun signaling pathway. However, it is unclear how DPP4 activates AKT signaling.
Integrins are a major family of cell surface receptors, which can activate intracellular signaling pathways including the FAK/AKT pathway that control tumor metastasis. 36,37 As a transmembrane protein, DPP4 has been demonstrated to be related to integrin β1. 38 DPP4 knockdown decreased the phosphorylation of integrin β1 at Ser785, which is key for integrin β1 binding to extracellular matrix. 39 The present study demonstrated that DPP4 interacted with and stabilized the integrin α4β1 complex to phosphorylate FAK, thereby activating the AKT cascade.
Based on our findings, we proposed a model to clarify how DPP4 promotes thyroid cancer cell metastasis (Fig. 7). Briefly, increased expression of DPP4 activates the FAK/AKT pathway by interacting with and stabilizing integrin α4β1 complex. Activated AKT phosphorylates c-Jun and promotes its nuclear localization, thereby inducing TGF-β1 transcription, while secreted TGF-β1 promotes EMT process and the subsequent thyroid cancer cell metastasis by binding its receptor. However, inhibition of DPP4 by sitagliptin had no effect on this process, suggesting that DPP4-induced α4β1 integrin activation is independent of its enzymatic activity.
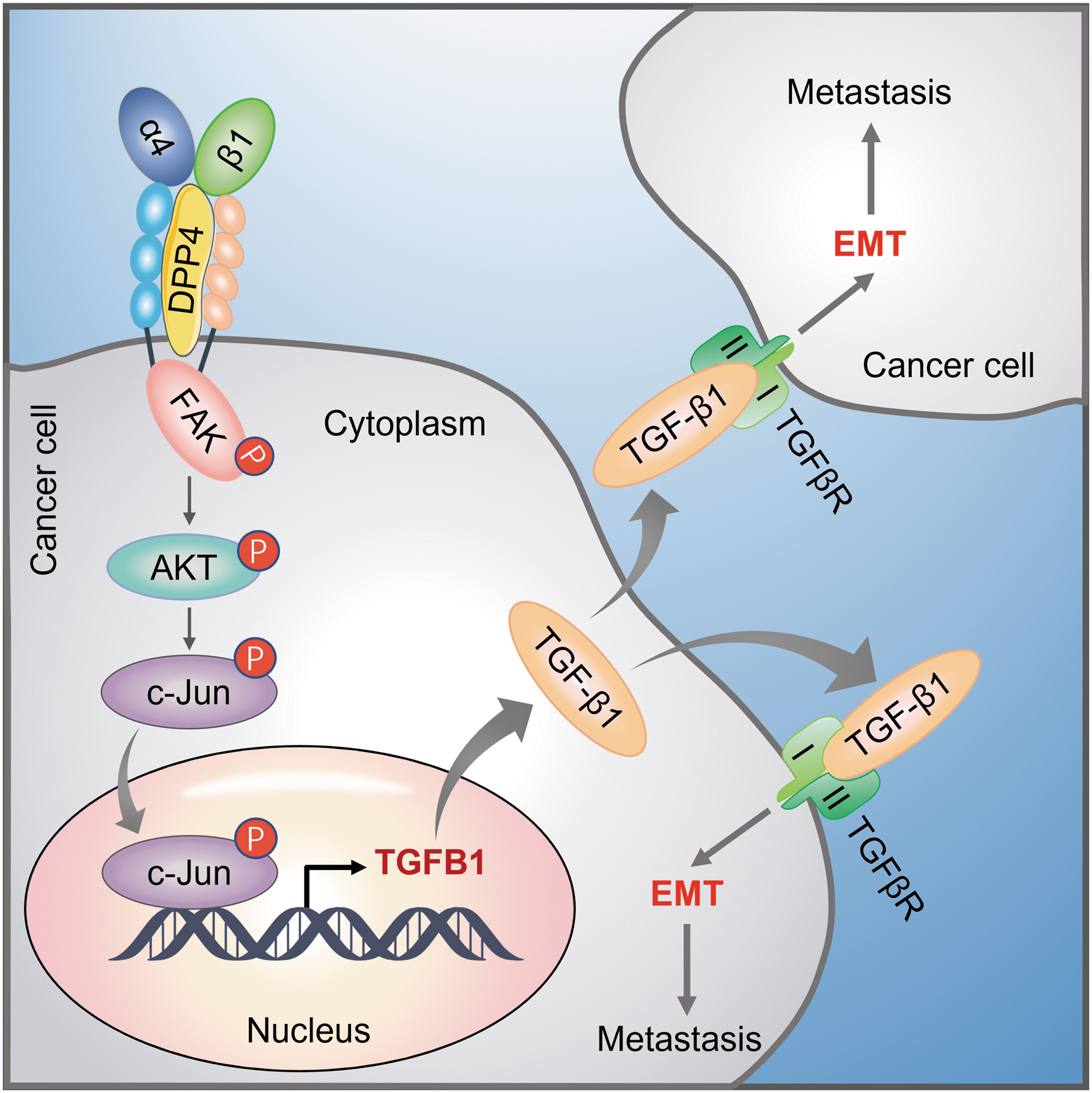
A schematic model of DPP4 promoting thyroid cancer metastasis by the integrin/FAK/AKT/c-Jun signaling axis. In thyroid cancer cells, DPP4 interacts with and stabilizes the integrin α4β1 complex, thereby activating the FAK/AKT signaling pathway. Activated AKT phosphorylates c-Jun and promotes its nuclear localization, where c-Jun induces TGFB1 transcription. Then, secreted TGF-β1 induces EMT and promotes tumor metastasis by activating its downstream signaling pathways.
In addition to DPP4 inhibitors, there are also other strategies to target DPP4, such as monoclonal antibodies and therapeutic vaccines. 40 –43 Moreover, therapeutic peptides are usually designed to block protein–protein interactions. 44,45 Thus, more studies are needed to develop new strategies for the treatment of thyroid cancer by blocking the interaction between DPP4 and α4β1 integrins.
Footnotes
Acknowledgments
We appreciate Dr. Haixia Guan (Guangdong Provincial People's Hospital, Guangzhou, P.R. China) for kindly providing the human thyroid cancer cell lines BCPAP, 8305C, and IHH4. We also would like to thank Drs. Kimura Shioko (National Institutes of Health, USA) and Martin McMahon (University of California, USA) for kindly providing the transgenic mouse strains TPO-Cre and BrafCA.
Authors' Contributions
M.J., X.Z., and P.H. contributed to the conception and design of the study. Q.H., Y.Z., and H.C. collected the experimental data. P.C., R.C., and N.W. collected tissue samples. Q.H. and W.L. contributed to the data analysis. P.H., Q.H., and W.L. wrote and revised the article. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (No. 82172675 and 81972593), and the Postdoctoral Innovation Base of Xi'an Central Hospital (No. 2020XASRSJ14).
Supplementary Material
Supplementary Data