
Abstract
Background:
In Down syndrome (DS), there is high occurrence of congenital hypothyroidism (CH) and subclinical hypothyroidism (SH) early in life. The etiology of CH and early SH in DS remains unclear. Previous research has shown genome-wide transcriptional and epigenetic alterations in DS. Thus, we hypothesized that CH and early SH could be caused by epigenetically driven transcriptional downregulation of thyroid-related genes, through promoter region hypermethylation.
Methods:
We extracted whole blood DNA methylation (DNAm) profiles of DS and non-DS individuals from four independent Illumina array-based datasets (252 DS individuals and 519 non-DS individuals). The data were divided into discovery and validation datasets. Epigenome-wide association analysis was performed using a linear regression model, after which we filtered results for thyroid-related genes.
Results:
In the discovery dataset, we identified significant associations for DS in 18 thyroid-related genes. Twenty-one of 30 significant differentially methylated positions (DMPs) were also significant in the validation dataset. A meta-analysis of the discovery and validation datasets detected 31 DMPs, including 29 promoter-associated cytosine–guanine dinucleotides (CpG) with identical direction of effect across the datasets, and two differentially methylated regions. Twenty-seven DMPs were hypomethylated and promoter associated. The mean methylation difference of hypomethylated thyroid-related DMPs decreased with age.
Conclusions:
Contrary to our hypothesis of generalized hypermethylation of promoter regions of thyroid-related genes—indicative of epigenetic silencing of promoters and subsequent transcriptional downregulation, causing biochemical thyroid abnormalities in DS—we found an enrichment of hypomethylated DMPs annotated to promoter regions of these genes. This suggests that CH and early SH in DS are not caused by differential methylation of thyroid-related genes. Considering that epigenetic regulation is dynamic, we hypothesize that the observed thyroid-related gene DNAm changes could be a rescue phenomenon in an attempt to ameliorate the thyroid phenotype, through epigenetic upregulation of thyroid-related genes. This hypothesis is supported by the finding of decreasing methylation difference of thyroid-related genes with age. The prevalence of early SH declines with age, so hypothetically, epigenetic upregulation of thyroid-related genes also diminishes. While this study provides interesting insights, the exact origin of CH and early SH in DS remains unknown.
Introduction
Down syndrome (DS) is a common chromosomal aneuploidy affecting around 1 in 750 live births. The DS phenotype includes a broad range of clinical features, including characteristic dysmorphism, short stature, congenital heart defects, intellectual disability, Alzheimer's disease, and thyroid dysfunction. 1
While more than 50 years have passed since the discovery of the cause of DS—the presence of a part of or a complete supernumerary chromosome 21—the exact pathophysiological mechanisms underlying DS-related phenotypes remain vague. Recent breakthroughs in DS research, however, have taught us that aneuploidy of chromosome 21 results in genome-wide epigenetic alterations. 2 –4
Epigenetics refers to the dynamic process of reversible heritable changes to gene expression, which is affected by the environment and genetics. Mechanisms include DNA methylation (DNAm; the most extensively studied mechanism), histone modification, and noncoding RNA-associated gene silencing. DNAm occurs at cytosine–guanine dinucleotides (CpGs) in the linear DNA sequence, where a methyl group can be transferred to cytosine at the C5 position, forming 5-methylcytosine.
DNAm is associated with gene expression in several ways, for example, by silencing gene promoter regions. 5 Chromosome 21 encodes multiple genetic factors that influence epigenetic transcriptional regulation (such as DNAm), and possibly due to the dosage effect of these genetic factors in DS, expression of a large number of genes on all chromosomes is dramatically changed. 3,4,6
The thyroid phenotype of DS is characterized by a higher incidence of congenital hypothyroidism (CH) and acquired autoimmune thyroid dysfunction. 7 –10 Besides these clinically evident forms of thyroid dysfunction, a high prevalence of nonautoimmune subclinical hypothyroidism (SH) has been observed in children—in up to 80% of young infants, decreasing to around 20% and 10%, respectively, in children and teens with DS. 11 –13
In addition, the mean size of thyroid glands is lower in children with DS. 14 A better understanding of CH and early SH in DS is needed for optimal decision-making with respect to thyroid hormone treatment of children with DS.
In newborns with DS, the Gaussian distribution plot of blood thyroxine (T4) concentration is shifted to the left and the distribution plot of serum thyrotropin (TSH) is shifted to the right. 7,11 This group phenomenon and the aforementioned lower thyroid gland size in children with DS may point to a mild to moderate form of DS-specific primary CH.
Since pathogenic variants in thyroid-specific genes are rarely found in DS, 9,15 we hypothesized that CH and early SH in DS may be caused by epigenetically driven gene expression alterations in genes controlling thyroid gland development and function, or genes involved in thyroid hormone action or metabolism. We further hypothesized that these are possibly alterations leading to hypermethylation of promoter regions of these genes and subsequently loss of function due to decreased expression.
In this study, we analyzed four DNAm datasets of individuals with DS compared with non-DS individuals, with a specific focus on thyroid-related genes, to gain insights into the molecular mechanisms underlying CH and early SH in DS.
Materials and Methods
Study data
Four DNAm datasets were included, comparing a total of 252 individuals with DS with 519 individuals without DS (Table 1). 16 –19 For comprehensive DNA extraction protocols, we refer to the original publications. 16 –19 In all four studies, DNA was extracted from whole blood samples.
Blood DNA Methylation Datasets Included in This Study
DNAm, DNA methylation; DS, Down syndrome; HM450, Illumina Infinium HumanMethylation450K BeadChip array; HM850, Illumina Infinium MethylationEPIC BeadChip array.
Statistics
A full description of the epigenome-wide association study (EWAS) analyses that we performed is given in Supplementary File S1. In short, original raw IDAT files—DNAm data derived from whole blood DNA analyzed with a DNAm array—were analyzed in R (v4.1.0) using the R package, SEnsible Step-wise Analysis of DNA MEthylation BeadChips (SeSAMe), 20 after which we used a linear regression model to analyze (1) the Muskens et al dataset (196 DS and 439 non-DS individuals; the discovery dataset due to its high number of participants) and (2) the Naumova et al, Henneman et al, and Bacalini et al datasets combined (54 DS and 80 non-DS individuals; the validation dataset).
The EWAS outcomes were filtered to only include CpGs that map within a ±2000-bp region of our thyroid-related gene panel of 49 genes (Supplementary Table S2). This gene panel is based on the Amsterdam UMC, location AMC, next-generation sequencing diagnostic panel for several thyroid conditions. 21 Differentially methylated positions (DMPs) were defined as CpGs with a false discovery rate (FDR) p < 0.05. p values of both analyses were combined in a meta-analysis.
Ethics
Institutional review boards approved of the original experiments by Muskens et al 16 (California Health and Human Services Agency, University of Southern California, and University of California Berkeley Institutional Review Board [#2018-103]); Naumova et al 17 (Saint Petersburg State University Research Ethics Board [Ref 02-119]); Henneman et al 18 (Academic Medical Center Research Ethics Board [Ref NL45496.018.13]); and Bacalini et al 19 (S. Orsola Hospital Ethics Committee, University of Bologna [Ref #126/2007/U/Tess]).
Participants or parents/guardians of participants provided informed consent, except in the Muskens et al study, in which deidentified newborn dried blood spots were obtained from the California Biobank Program (SIS request nos. 572 and 600) with a waiver of consent from the Committee for the Protection of Human Subjects of the State of California. The research was completed in accordance with the Declaration of Helsinki as revised in 2013.
Results
Figure 1 shows a flow chart of the analysis with summarized findings.
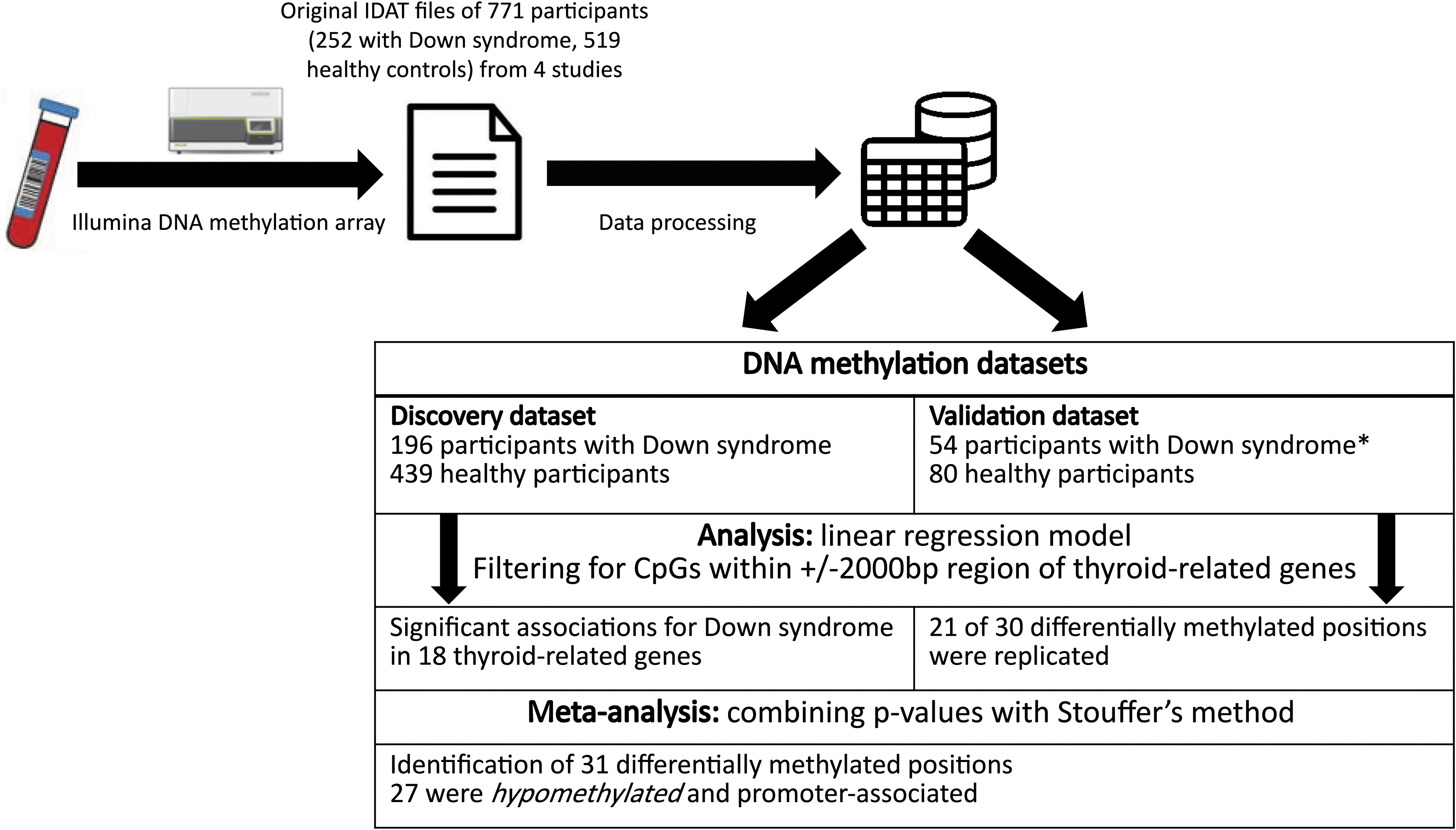
Overview of analysis and summarized findings. *Data of 2/56 participants with DS in the validation dataset were excluded due to more than 5% CpGs missing after data processing. CpG, cytosine–guanine dinucleotide; DS, Down syndrome.
Discovery set
To gain insights into the molecular mechanisms underlying CH and early SH in DS, we filtered the Muskens et al EWAS results for all CpGs within a ±2000-bp region of each gene in a thyroid-related gene panel (Supplementary Table S2), including 49 known genes implicated in thyroid gland development and thyroid hormone production, action, and metabolism. A total of 1645 (HM850) CpGs in autosomal genes were identified.
Differential methylation analysis revealed 54 DMPs associated with DS (Supplementary Table S3). A separate association analysis was conducted for CpGs annotated to the allosomes, segregating males and females (Supplementary File S1). This analysis did not identify CpGs that reached statistical significance in males, whereas in the female-stratified analysis, we detected two DMPs in TBL1X (Supplementary Table S4).
Of the 54 significant autosomal DMPs (Supplementary Table S3), 34 (63%) showed any level of hypomethylation—a negative delta β (measure of methylation) in DS patients compared with controls—and 20 (37%) showed hypermethylation. According to the GENCODE genomic annotation, 45 DMPs were associated with a gene promoter region (defined by annotation of the CpG to the 5′UTR, the 1500-bp range upstream the transcription start site, or the first exon of a gene).
There were 10 genes harboring one significant DMP and 8 genes with multiple significant DMPs, including 2 genes—GNAS and DYRK1A—with 15 and 12 significant DMPs, respectively. It should be noted that 29 GNAS CpGs and 8 DYRK1A CpGs were involved in 4 significant differentially methylated regions (DMRs) identified in the original hypothesis-free (epigenome-wide) analysis of the Muskens et al dataset 16 : hg 19 chr20:57426858-57427977 (GNAS) and hg19 chr21:38737243-38737860, chr21:38738865-38739180, and chr21:38807423-38807712 (all DYRK1A).
Validation set and meta-analysis of thyroid-related DS DNAm data
To explore the consistency of our results, we revisited three smaller DS DNAm datasets containing samples of three distinct DS and control populations: toddlers, newborns, and adolescents/adults. First, the findings of the discovery analysis were validated by analyzing the validation DNAm datasets in a similar manner as the discovery set.
Subsequently, we inspected the consistency of our results considering age differences between the four datasets (see the “Effect of age on methylation of thyroid-related genes” section). A t-distributed stochastic neighbor embedding (t-SNE) plot for data exploration, including the discovery set and validation sets, showed some clustering of samples according to the study and age group; however, scattering of samples due to batch effects and ancestry was also clearly visible (Fig. 2A, B).
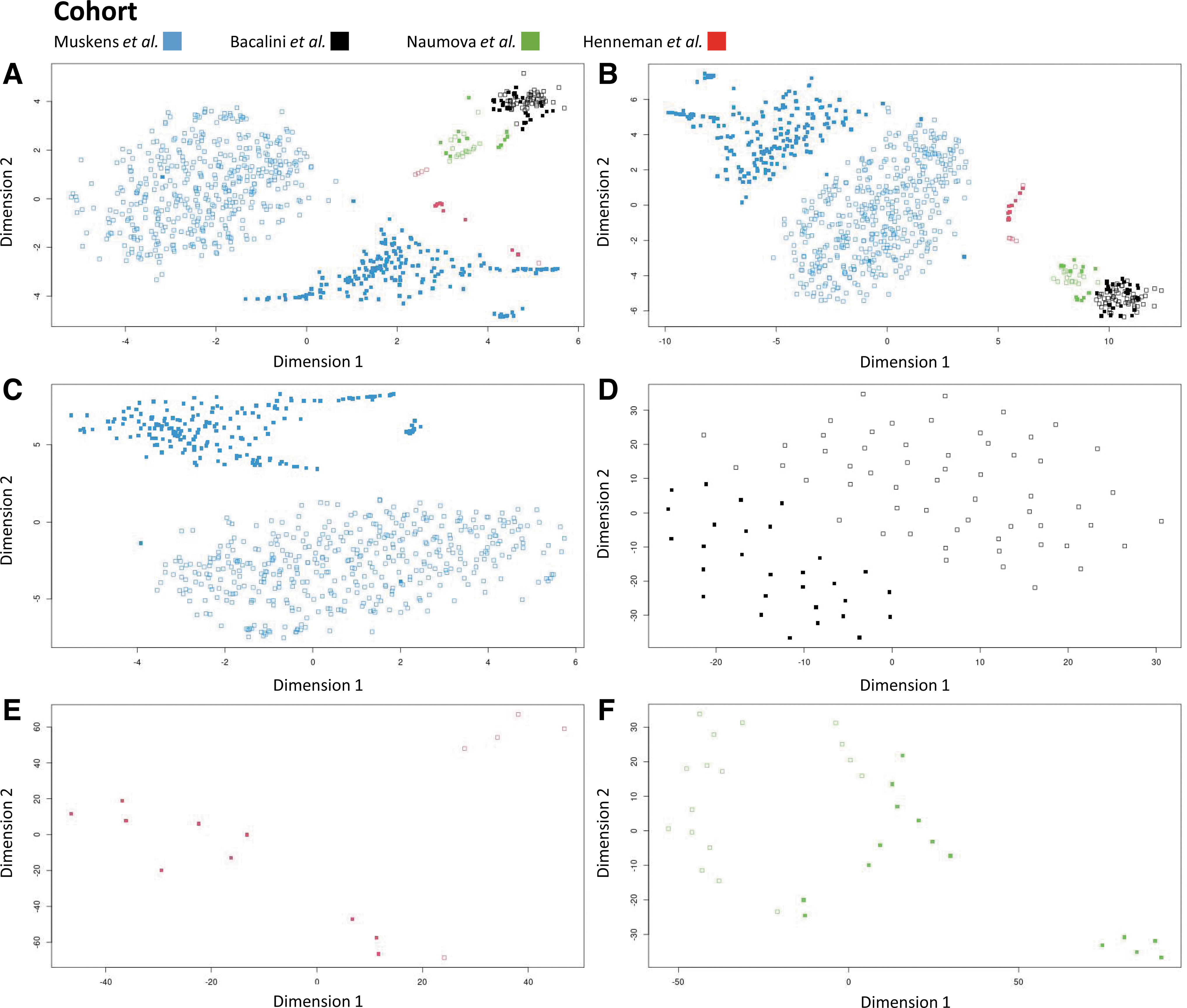
t-SNE plot of DNAm data in individuals with DS and non-DS individuals. t-SNE is a statistical method for visualizing high-dimensional data. In the t-SNE scatterplot, similar participants are modeled by nearby dots and dissimilar participants are modeled by distant dots. Filled-in squares represent individuals with DS, and empty squares represent non-DS individuals.
t-SNE plots of individual datasets showed clear segregation of DS patients and controls (Fig. 2C–F); these case–control differences were also observed in the thyroid-critical CpGs (Fig. 2B).
We applied the Benjamini–Hochberg FDR correction to p values of two subsets of CpGs resulting from the validation set, EWAS: (1) the list of 54 significant DMPs detected in the discovery analysis and (2) all 1645 CpGs that were analyzed in the discovery analysis. The first and second subsets of CpGs in the aggregate validation set consisted of 30 and 949 CpGs, respectively, due to differences between HM850 and HM450 DNAm arrays (Table 1).
In the first subset of CpGs (Supplementary Table S3), 25 of the 30 DMPs showed the same direction of methylation change between DS and non-DS groups across the datasets and 21 of 30 were statistically significant in the validation dataset. Most of these DMPs were located within the GNAS gene; 13 of 14 GNAS DMPs present in the HM450 methylation array were statistically significant in the validation dataset.
In the second subset of CpGs, we performed a meta-analysis based on p values of the discovery and validation sets (Supplementary Table S5). The meta-analysis detected 31 DMPs, including 29 promoter-associated CpGs with identical direction of effect across the discovery and validation sets and a combined p < 0.05 in the GNAS, HOXA3, DYRK1A, PAX8, THRB, DUOX2, TBX1, TPST2, TG, and TUBB1 genes. Nine DMPs mapped to CpG islands and 12 to CpG island shores.
Notably, 29 of 31 DMPs were hypomethylated in the DS group. To investigate if there was an enrichment of hypomethylated DMPs, we compared the number of hypomethylated versus hypermethylated DMPs that were significant in the meta-analysis with CpGs that were not significant in the meta-analysis, and the difference was highly significant (Fisher's exact test p < 0.0001). Of all thyroid-related genes, GNAS was again the most prevalently associated gene with DS, further strengthening the association of GNAS differential methylation with DS (Table 2).
Differentially Methylated Positions Detected in the Meta-Analysis Near/in Genes Associated with Thyroid Gland Development and Thyroid Hormone Production, Action, and Metabolism (Down Syndrome Vs. Non-Down Syndrome)
Furthermore, two of four DMRs in thyroid-related genes reported in the original analysis of the Muskens et al dataset 16 were replicated in the meta-analysis (Fig. 3): the GNAS DMR, hg19 chr20:57427472-57427830, containing 12 CpGs (due to differences between HM850 and HM450 DNAm arrays) with a mean β difference of −0.082 and one (of three) DYRK1A DMR, hg19 chr21:38807423-38807712, containing two CpGs with a mean β difference of 0.0923 (Šidák-corrected p = 4.78E-03 and 2.09E-03, respectively).
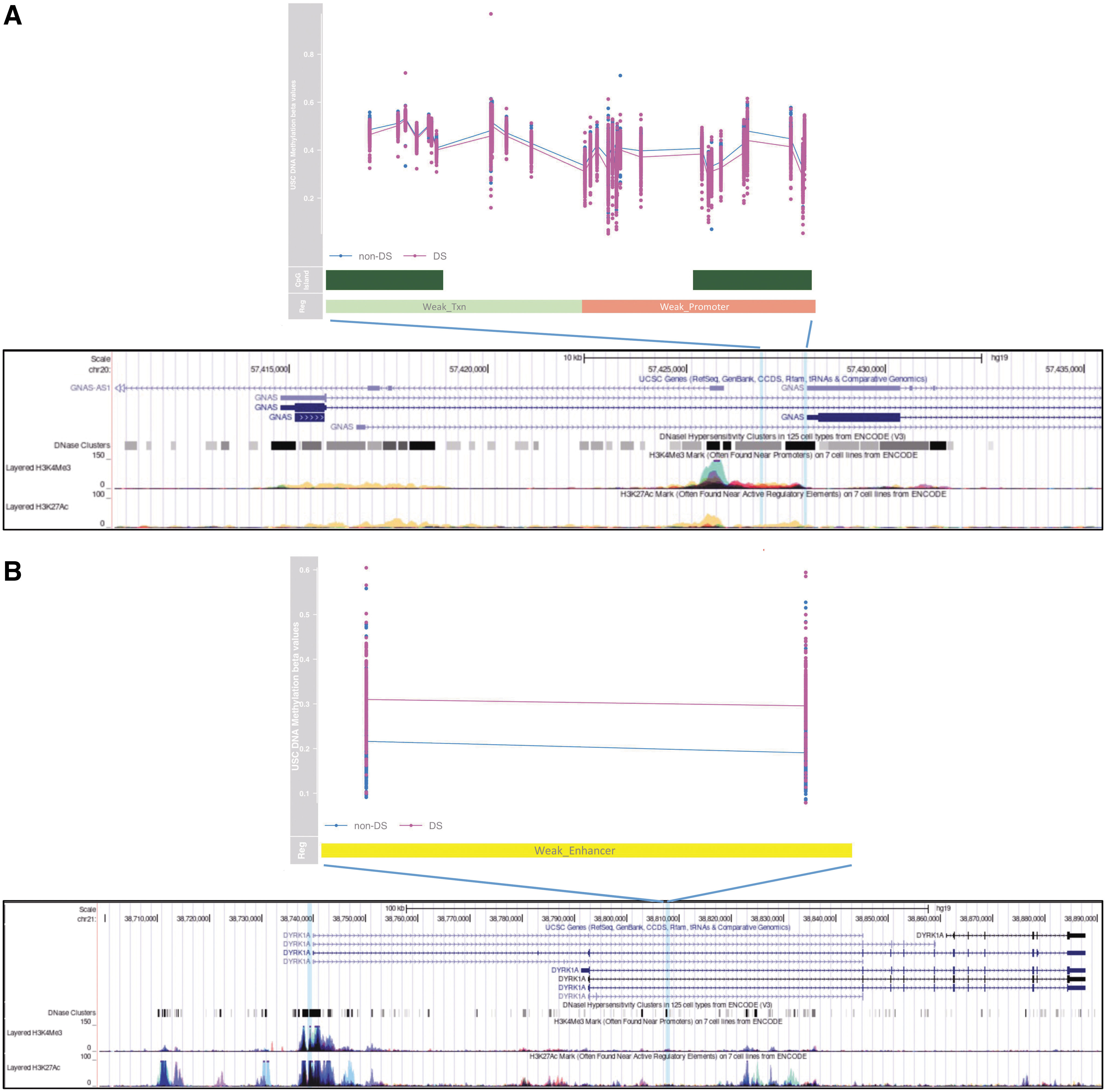
GNAS and DYRK1A DMRs. GNAS and DYRK1A DS-associated DMRs (hg19) identified in the original hypothesis-free analysis (without applying thyroid-related gene set filtering) of the Muskens et al dataset—including DNAm profiles of DS (purple, N = 196) and non-DS (blue, N = 439) newborns.
16
Mean delta β values of DMRs were
The validation set association analysis for CpGs on the allosomes, stratified by sex, revealed comparable results as in the discovery set. Considering CpGs that are present in both HM850 and HM450 data, no significant DMPs were detected (Supplementary Table S4).
Effect of age on methylation of thyroid-related genes
In children with DS, the prevalence of SH and low serum T4 decreases with age. 10,12,22 It is conceivable that differential DNAm of thyroid genes over time is involved in this process. To shed light on this hypothesis, we explored the change in mean delta β values of DMPs detected in the meta-analysis across the DNAm datasets/age groups (for comprehensive methods, see Supplementary File S1).
The Muskens et al and Henneman et al data were combined to form the age group “newborns,” and the Naumova et al and Bacalini et al data constituted the “toddlers” and “adolescents/adults” groups, respectively. Delta β was computed by linear regression, adjusting for cell type proportions.
We found that the mean delta β of hypomethylated thyroid-related DMPs (29 of 31 DMPs) decreases with age (Fig. 4). In other words, there is a smaller methylation difference between DS patients and controls at an older age for most of the thyroid-related DMPs associated with DS. An ANOVA of mean delta β values of the three age groups indicated a significant difference (F-value: 271.3 [critical F-value = 3], p = <0.0001).

Age-stratified analysis of DMPs identified in the meta-analysis. Colored dots indicate mean β values of DMPs identified in the meta-analysis, stratified per age group. Trajectories are highlighted by dashed lines. Both hypomethylated and hypermethylated DMPs are shown. Black dots represent mean delta β values (± pooled standard errors) of all hypomethylated DMPs, stratified per age group. DMP, differentially methylated position.
The hypermethylated DMPs—two of 31, specifically cg21505925 and cg19988492, which are both associated with the DYRK1A promoter region—showed a trend toward an increase in differential methylation between newborns/toddlers and adults (Fig. 4).
Discussion
Investigation of patterns of aberrant DNAm in DS may provide insight into certain aspects of the DS phenotype. We hypothesized that the molecular mechanism of CH and early SH in DS may involve epigenetic changes to thyroid-related genes, resulting in their transcriptional deregulation. To explore this, we revisited blood DNAm profiles of individuals with DS and individuals without DS in four separate datasets. 16 –19 DMPs and DMRs associated with DS in several thyroid-related genes were detected in both discovery and validation datasets.
Contrary to our expectation of generalized hypermethylation of promoter regions of thyroid-related genes—indicative of epigenetic silencing of promoters and subsequent transcriptional downregulation of these genes, causing biochemical thyroid abnormalities in DS—we found an enrichment of hypomethylated DMPs annotated to promoter regions of these genes. This suggests that CH and early SH in DS are not caused by differential methylation/expression of thyroid-related genes.
In the discovery analysis, where we filtered the results of a large EWAS of DS newborns for CpGs of thyroid-related genes (Supplementary Table S2), 16 we mostly identified hypomethylated DMPs associated with GNAS and hypermethylated DMPs associated with DYRK1A. We validated the results in an aggregate dataset comprising three smaller DS DNAm datasets and replicated a substantial amount of DMPs, and we showed that the direction of effect was similar for almost all these DMPs (Supplementary Table S3).
A meta-analysis of the discovery set and validation set indicated that of the 49 thyroid-associated genes, 10 genes showed a DS-specific methylation profile (Table 2), in most cases, related to promoter region hypomethylation. In addition, the meta-analysis again showed that there is an enrichment of significant GNAS DMPs.
In view of GNAS, we found an association between DS and hypomethylation of the promoter of a paternally expressed GNAS transcript, XLαs. XLαs is the large variant of the stimulatory G protein α subunit (Gsα), which is important for cellular signaling in the context of several hormone systems, such as the TSH signaling pathway. 23,24
Methylation defects of several GNAS DMRs, including the XLαs promoter region, are associated with pseudohypoparathyroidism, 25 a condition characterized by resistance to multiple stimulatory G protein-coupled hormones. It is difficult to decipher the clinical implication of isolated hypomethylation of the XLαs promoter in the context of CH and early SH in DS; however, we speculate that it may indicate an epigenetic compensation effect consequent to thyroid dysfunction.
While XLαs is normally expressed at low levels in the thyroid, 24 hypomethylation of its promoter could signal upregulation to counter CH and early SH in DS. Future studies of XLαs expression and methylation in DS are likely to clarify this phenomenon, which could also provide further insight into how XLαs and Gsα expression is regulated.
Reduced expression of all genes with hypomethylated DMPs identified in the meta-analysis (except for DIO1) is associated with CH in humans. 26,27 The fact that we almost exclusively identified hypomethylation of promoter-associated DMPs strengthens our hypothesis of an epigenetic rescue phenomenon, bearing in mind the notion that DS was characterized by significant genome-wide hypermethylation in a number of studies. 16,17,19,28 –30
Thus, these findings may suggest that CH and early SH in DS do not result from epigenetic alterations of thyroid-related genes, but that CH and early SH may influence the formation of these alterations. Although the cause of CH and early SH in DS still remains to be elucidated, it may be derived from several DNAm and transcriptomic analyses: it was reported that deregulated genes in DS are associated with basal biological processes such as cell development, cell cycle, and tissue morphogenesis. 16,17,31,32
To develop a full picture of CH and early SH in DS and to validate the presented DNAm associations, additional studies will be needed, which investigate gene expression and DNAm in thyroid tissue. Recently, the transcriptomic signature of fetal thyroid tissue has been studied for the first time. 33 Similarly, future work could be carried out to study DS fetal thyroid tissue.
Considering DYRK1A, we found hypermethylation of promoter-associated DMPs. The increased expression of DYRK1A (a chromosome 21 gene) due to the dosage effect has been suggested as a potential causative mechanism of the observed thyroid hypoplasia in DS. 34 A Dyrk1a transgenic mouse model that overexpressed Dyrk1a showed a significant decrease of T4 and morphological impairment of thyroid tissue. 34
However, in humans, DYRK1A has not yet been associated with thyroid dysfunction. In case DYRK1A is also implicated in thyroid function in humans, the observed hypermethylation of the DYRK1A promoter may also be interpreted as an epigenetic rescue phenomenon to ameliorate CH and early SH phenotypes of DS since hypermethylation of the promoter region suggests transcriptional downregulation.
Nonautoimmune SH in DS shows a trend of declining prevalence with age. 10,12,22 A possible explanation for this phenomenon may be maturation of the thyroid gland and a decreasing need for the thyroid hormone with aging, which is commonly observed in some monogenic forms of mild thyroidal CH. 35 A recent meta-analysis of DNAm of individuals with DS compared with non-DS individuals in the Muskens et al, Naumova et al, and Bacalini et al datasets showed a clear trend across different age groups. 17
The methylation difference, or delta β, in both hypomethylated and hypermethylated DMPs was greater in older individuals with DS. We replicated this analysis and found contrasting results concerning hypomethylated thyroid-related DMPs, where delta β differences between DS patients and controls seemed to be inversely correlated with age (Fig. 4): the methylation difference was greater in younger subjects than in older subjects. This observation aligns with the hypothesis that the thyroid gene methylation differences could be rescue effects consequent to thyroid dysfunction.
Hypothetically, because of the declining prevalence of nonautoimmune SH with age, the need for methylation-related upregulation of thyroid genes also diminishes. Future longitudinal methylation studies in DS, which also take thyroid hormone and TSH levels into account, will need to be undertaken to explore this idea.
Our work presents several limitations. First, the main results of this study stem from a candidate gene analysis of DNAm data, and conclusions drawn from the analysis rely on biological inference. Of note, not much is known about the interplay between DNAm and transcriptional regulation in (healthy) thyroid tissue. Hence, we are not sure whether the promoter regions containing the DS-associated DMPs are epigenetically active in thyroid tissue.
Second, the findings of this study may be somewhat limited by the use of DNAm patterns derived from blood. While blood DNAm can be used as a proxy for DNAm of other tissues, 4,30 the best results are accomplished by studying the tissue of interest—in this case, thyroid tissue.
Last, as expected, batch effects and subject heterogeneity posed a problem to the combined analysis of different datasets. Their effect on the data may even be as large as the influence of DS, as observed in Figure 2A and B.
However, when inspecting individual datasets separately, DS remains the main factor driving variation in DNAm between samples (Fig. 2C–F), including among the thyroid gene-related CpGs (Fig. 2B). Therefore, to solve this problem, study and subject age were added as variables in the analysis of the validation set.
A strength of our work lies in the methodology of our validation analysis, where we applied the same methods for preprocessing and the same parameters for analysis as in the discovery analysis. In addition, by aggregating the smaller datasets and performing a meta-analysis, we acquired 80% power to reliably report on a delta β value of around 2% (β difference of 0.02 or −0.02) or more at critical α = 0.05. 36
In summary, the (nonautoimmune) thyroid phenotype of children with DS was characterized in several studies by increased incidence of CH, increased prevalence of SH, and smaller thyroid glands. Moreover, taken as a group, persons with DS have abnormal biochemical indices of thyroid function compared with healthy people. In light of the recently discovered genome-wide transcriptional and epigenetic alterations in DS, we hypothesized that CH and early SH in DS may be caused by promoter region hypermethylation of thyroid-related genes.
In contrast, we mainly found promoter region hypomethylation, signaling transcriptional upregulation. This suggests that CH and early SH in DS are not caused by differential methylation/expression of genes involved in thyroid development or function. Since epigenetic regulation is dynamic by definition, we may speculate that the identified changes to DNAm of thyroid-related genes could be a rescue phenomenon to ameliorate the nonautoimmune thyroid phenotype.
While this study provides interesting insights, the exact origin of CH and early SH in DS remains unknown. Focus of further research should be centered around obtaining and analyzing DS thyroid tissues and thyroid hormone target tissues and age-matched controls for (multi)-omics analyses.
Footnotes
Acknowledgment
The authors are thankful for the data on all subjects in the different studies used in this work.
Authors' Contributions
P.L. was involved in writing—original draft, statistical analyses, and conceptualization; N.Z.-S. was involved in writing—original draft, review, and editing; S.L. was involved in review and editing and statistical analyses; M.G.B. and O.Y.N. were involved in review and editing, acquiring data, and revision of intellectual content; J.W. and A.B. were involved in review and editing and revision of intellectual content; P.H. and A.J.d.S. were involved in review and editing, acquiring data, project coordination, statistical analyses, and conceptualization; and A.S.P.v.T. was involved in writing—original draft, review and editing, project coordination, and conceptualization. All authors critically reviewed manuscript drafts and approved the final manuscript as submitted.
Availability of Data and Materials
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary File S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5