
Abstract
Background:
Congenital hypothyroidism due to defects in iodotyrosine deiodinase has variable phenotypes and can present as hypothyroid or with normal thyroid testing.
Methods:
Whole exome sequencing was performed in individuals from two families originating from different regions of Sudan. Mass spectrometry of urine and serum iodotyrosines was performed on subjects from both families.
Results:
A novel iodotyrosine deiodinase (IYD) mutation (c.835C>T; R279C) was identified in individuals from two Sudanese families inherited as autosomal recessive. The mutation was identified by multiple in silica analyses to likely be detrimental. Serum and urine monoiodotyrosine (MIT) and diiodotyrosine (DIT) were markedly elevated in the homozygous subjects.
Conclusion:
Measurement of serum and urine DIT and MIT was more sensitive than that of urine iodine or serum thyroid function tests to determine the effect of the IYD mutation.
Introduction
Congenital hypothyroidism (CH) affects 1:3000 newborns. A delay in diagnosis and treatment may result in significant growth and developmental retardation as well as severe intellectual impairment. Thyroid dysgenesis is the cause of 65% of CH, while dyshormonogenesis and gland in situ accounts for 35% of CH. 1 Iodine is an essential component of thyroid hormone (TH) synthesis and when iodine is limited in supply, TH synthesis is impaired. Assurance of sufficient supply of iodine for TH synthesis depends on (1) the environmental supply of iodine, (2) the ability of the thyroid to transport iodine into the thyroid cells, (3) sufficient recycling of iodotyrosines so that maximum efficiency of iodine utilization occurs.
Multiple genes are involved in TH synthesis and specifically iodine metabolism. 2 Release of thyroxine (T4) and triiodothyronine (T3) from the thyroid gland requires proteolysis of thyroglobulin, during which monoiodotyrosine (MIT) and diiodotyrosine (DIT) are also released. The iodine of MIT and DIT cannot be used directly and require deiodination by the enzyme iodotyrosine deiodinase (IYD). IYD allows iodine to be recycled to synthesize more T3 and T4. 3 When environmental iodine is not in abundance, the presence of functional IYD becomes more important in conservation and utilization of iodine in the synthesis of TH.
We report two Sudanese families from different parts of the country with CH and goiter due to a novel mutation in the IYD gene. The phenotype as determined by thyroid function tests and goiter were variable ranging from hypothyroid to euthyroid despite the same genetic defect in the two families. Whereas urine iodine concentration was unable to predict the degree of iodine deficiency in these patients, liquid chromatography coupled to tandem mass spectrometry analysis of serum and urine confirmed excessive accumulation of MIT and DIT in the affected patients. These families illustrate the utility of measurement of MIT and DIT in diagnosing CH in iodine-sufficient and iodine-borderline areas.
Methods
All patients were referred to a pediatric endocrinologist at the University of Khartoum, Sudan, presenting with stigmata of hypothyroidism or due to a sibling having hypothyroidism. Oral consent from patients or their guardians and their family members was obtained before blood sampling due to literacy. Studies were approved by the University of Miami Institutional Review Board. The initial thyroid testing (thyrotropin [TSH] and free T4) was done in Sudan at the time of diagnosis. When the patients came for follow-up at their local clinic in Sudan, blood was then sent to Miami, Florida.
Testing was performed using the Immulite® 1000 (Siemens, Munich, Germany) platform as previously described. 4 Isolation of genomic DNA from whole blood using the Qiagen QIAamp® DNA Blood Mini Kit (Hilden, Germany) was carried out at the University of Miami. Blood samples were obtained from propositus, siblings and parents when possible. For each of the 2 families, gDNA from the proband along with 1 parent was submitted to whole exome sequencing (WES) (Novogene, Agilent SureSelect Human All Exon V6 Kit). A compilation of thyroid genes linked to thyroid disorders was evaluated and possible mutations linked to the phenotype were identified based on predicted functional scores, allele frequency, and zygosity. 4
Then gDNA from all family members available was analyzed and confirmed by Sanger sequencing (Genewiz, Abi 3730xl DNA Analyzer) to verify the WES results and establish the genotype of all sampled family members. All identified variants were further evaluated by in silico prediction scores. 5 –9
Measurement of MIT and DIT in serum and urine was performed by liquid chromatography coupled to tandem mass spectrometry. 10 Iodine was quantified by the Sandell–Kolthoff method. 11
Case Presentations and Results
Family 1
Family 1 (Fig. 1A) from western Sudan was identified in 2017 when the propositus, from consanguineous parents (first cousins), presented at age 7 years with impaired cognitive function, goiter, and found to have elevated TSH and low serum T4. He was started on levothyroxine (LT4) and prompted evaluation of a younger brother aged 8 years and a younger sister aged 3 years. Both siblings had what was considered to be “low IQ” by the clinician with goiter on examination, and thyroid function tests confirmed tests similar to propositus with high serum TSH and low serum T4 with classic hypothyroid facies of periorbital edema and swollen lips (Fig. 1B).

Pedigree, thyroid function tests, and urine iodine values of Family 1. (
Serum was obtained 7 days after starting LT4 therapy and again after 6 months and sent to Miami for further analysis (Fig. 1C). By 7 days of treatment, thyroid function tests of the propositus (II-1) and his brother (II-2) normalized, although those of the younger sister (II3) did not normalize until 6 months (Fig. 1A). WES carried out on the propositus and his mother demonstrated a mutation in exon 5 (c.835C>T; R279C) of the IYD gene. Sanger sequencing confirmed that all three siblings (II-1, II-2, and II-3) were homozygous for the mutation, while the parents were heterozygous (Fig. 1D).
Due to the discovery of the IYD gene mutation, LT4 supplementation was discontinued and the 3 siblings were treated with Lugol's iodine 2 drops twice daily for 4 weeks and repeated thyroid tests confirmed maintenance of euthyroidism (Supplementary Table S1). After four weeks, due to difficulty in obtaining Lugol's iodine, the patients were switched back to LT4 with continued maintenance of normal thyroid tests. Spot urine iodine was obtained after restarting LT4 (Fig. 1A).
Family 2
Family 2 from northern Sudan was identified in 2018 when the propositus (II-1, Fig. 2), born to consanguineous parents (first cousins), presented at age 7 and was placed on LT4. Available medical notes were incomplete. At age 13 years, he self-discontinued LT4, felt well, his thyroid function tests were normal, and he did not resume LT4 treatment. Siblings II-2 and II-3 had goiter and were not treated with LT4 but II-4 who did not have a goiter had some lethargy and was briefly treated with LT4. WES carried out on DNA from the propositus and mother demonstrated the same IYD mutation as that found in Family 1 (exon 5; c.835C>T; R279C) with subjects II-1, II-3, and II-4 homozygous, and the mother (I-2) and a sibling (II-2) heterozygous.
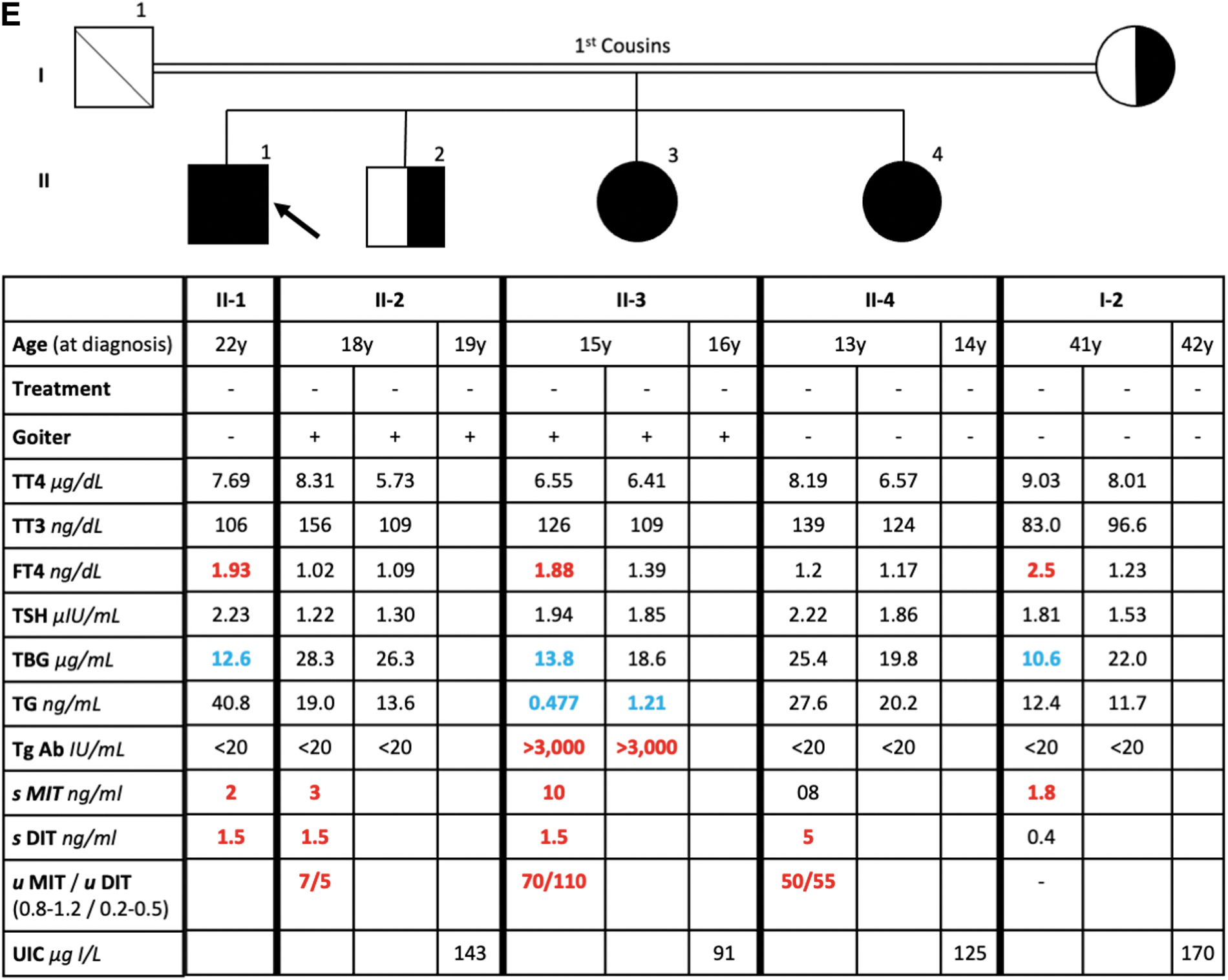
Pedigree, thyroid function tests, and urine iodine values of Family 2. Values in red are above the limit of normal and values in blue are below the limit of normal. Treatment a, 7 days after starting LT4; Treatment b, 6 months after starting LT4.
The father was deceased. Spot urine iodine was obtained (Fig. 2). Blood and urine were obtained on two different occasions and sent to Miami for analysis that showed similar normal values for all thyroid function tests and urine iodine.
Due to the identical mutation identified in these 2 families from different parts of Sudan, the gene locus was haplotyped demonstrating that the minimally shared genetic gene interval surrounding the mutation was 2.7 megabases (Supplementary Fig. 1).
Iodothyronine concentrations in serum and urine
Iodothyronine measurements were performed in serum from individuals of Family 1 (Fig. 1A) and urine and serum from individuals from Family 2 (Fig. 2). Serum MIT and DIT were markedly elevated in the serum of all homozygous individuals compared with the heterozygous father (I-1), and T3 and T4 concentrations were slightly higher in the affected subjects. In Family 2, serum MIT and DIT were markedly elevated in the affected individuals, whereas T3 and T4 were not. However, urine MIT and DIT were markedly elevated in the affected individuals.
Discussion
A novel mutation (c.835C>T; R279C) in the IYD gene was identified in two Sudanese families from different regions of Sudan, manifesting autosomal recessive inheritance. Multiple in silico analyses (SIFT, Polyphen2 HVAR, Polyphen 2 HDIV, Mutation Tester, Mutation Assessor, FATHMM and CADD) indicated that the mutation likely had a detrimental consequence.
The first reported patient with a dehalogenase defect, confirmed by measurement of iodotyrosines, was described in 1955. 12 The patient was born with goiter in an area of Holland where goiter was endemic. He was treated with “thyroid powder” at nine months of age that showed shrinkage of the goiter. A younger brother who was not treated with “thyroid powder” remained goitrous and mentally delayed. The parents were unrelated and had no goiter.
A series of elegant experiments demonstrated that intravenously administered 131I labeled dl-DIT and dl-MIT were excreted in the urine almost entirely unchanged. The authors concluded that there was a defect in dehalogenase. 13 While other cases were reported earlier, this was the first to directly demonstrate altered metabolism of iodotyrosines. Subsequently additional cases of presumptive dehalogenase defects were identified. 14
However, it was not until 50 years later when dehalogenase 1, or iodotyrosine dehydrogenase (IYD), was sequenced and characterized. 15 Since then, 4 unique IYD mutations have been reported in 9 individuals 16 –18 (Fig. 3). There is significant phenotypic variability in these patients even in those with identical mutations in different families as described in the present report with a novel fifth unique IYD mutation. TSH was variably elevated in 3 individuals before starting LT4 treatment (Patients 1, 4, and 8; Fig. 2), and in 1 individual (Patient 7, Fig. 3) the TSH ultimately became elevated 5 years after initial diagnosis.
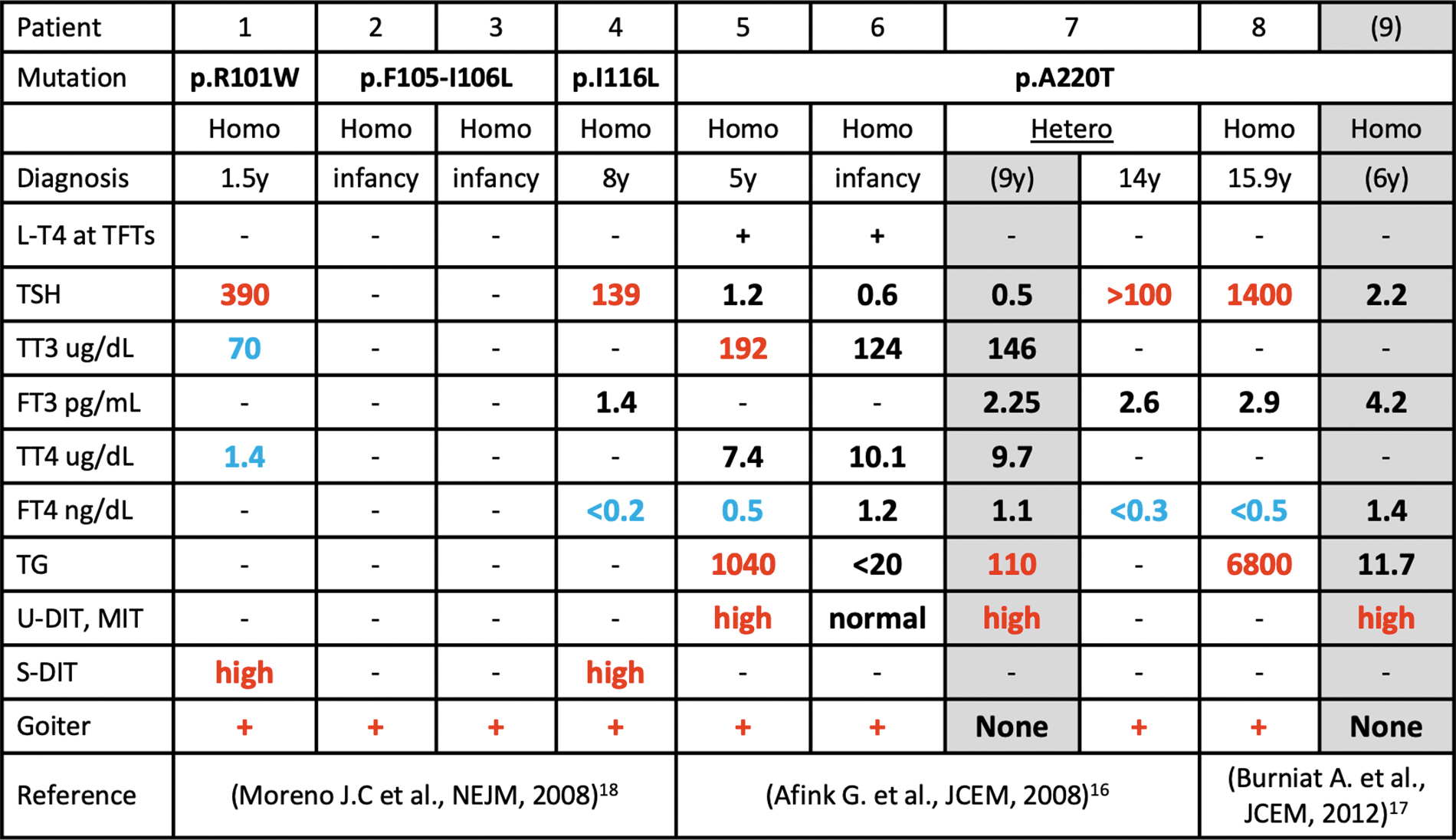
Comparison of phenotype and genotype in other families with IYD gene mutations. Values in red are above the limit of normal and values in blue are below the limit of normal. +, present; −, not performed or data not available.
Age at diagnosis and mode of inheritance were also variable (Fig. 2). MIT and DIT were consistently elevated. Loss of iodotyrosines in urine reduces the iodine available for recirculation, producing iodine deficiency. When the availability of environmental iodine is limited, loss of iodotyrosines lowers the threshold for hypothyroidism. To this point, in Family 1, replacement of iodine with Lugol's iodine solution was able to substitute for LT4 and maintain normal thyroid tests. In summary, measurement of serum and urine DIT and MIT was more sensitive than that of urine iodine or thyroid function tests to identify a defect in iodotyrosines deiodination and the presence of an IYD mutation.
Footnotes
Acknowledgments
We thank the patients for participation in the research and Dr. Gilbert Vassart for assistance in getting the samples from Sudan to Miami. Moreover, we thank the CISUP–Centre for Instrumentation Sharing of the University of Pisa for providing the Sciex QTrap 6500+ mass spectrometer, which was used for MIT and DIT assays. The authors thank Dr. Ali R. Mani for providing MIT and DIT standards.
Statement of Ethics
The authors have no ethical conflicts to disclose. The subjects agreed to participate in the study. The study was approved by the institutional review board at the University of Miami Miller School of Medicine protocol number 20140632
Authors' Contributions
R.S. contributed to identification of family in Sudan and collection of specimens; A.F. carried out haplotyping and review of the article; R.B. was involved in genotyping and identification of the gene in Family 2; Y.W. was in charge of genotyping and identification of the gene in Family 1; M.A.A. contributed to supervising collection of samples in Sudan; A.D. took care of review of article and intellectual input into the design of the study; S.R. took care of intellectual input into the design of the study and review of the article; M.B., A.B., and A.S. were in charge of mass spectrometry analysis; R.Z. was in charge of mass spectrometry analysis and intellectual input into the study and review of the article; R.E.W. conceived the study, gave intellectual input into design and raised funds for the study, and wrote the article.
Author Disclosure Statement
R.E.W. is on the advisory board and scientific cofounder for PriZm Therapeutics and on the advisory board of FBIIO Acquision Corp XXV. The other authors have no competing interests.
Funding Information
This study was supported by grants from the National Institutes of Health, USA, DK 15070 to S.R. and DK110322 to A.M.D., MD010722 to R.E.W. and by funds from the Esformes Thyroid Research Fund.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1