
Abstract
Background:
Treatment of Graves' hyperthyroidism (GH) and Graves' orbitopathy (GO) is far from adequate, and hence, new substances that specifically target the autoantigens in GH/GO are warranted. This study determined the preclinical in vitro efficacy of SYD5115, a novel low-molecular-weight compound that inhibits the thyrotropin receptor (TSH-R).
Methods:
The TSH-R inhibiting capability of SYD5115 was tested through stimulation of wild-type and chimeric TSH-R expressed in Chinese hamster ovary (CHO) cells using two functional (stimulatory and blocking) cell-based TSH-R-Ab bioassays. TSH-R expressing human orbital fibroblasts, collected from GH+GO patients (GOF), were stimulated with the monoclonal antibody M22 or with stimulatory TSH-R-Ab (TSAb)-positive sera with cyclic adenosine monophosphate (cAMP) or hyaluronic acid (HA) release as readouts. The effect of SYD5115 on the viability of GOF was tested in 4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide and scratch cell growth assays.
Results:
SYD5115 significantly and dose dependently inhibited the TSH-R activation through M22 or TSAb-positive sera in all performed bioassays. Inhibition showed similar levels in the TSAb reporter bioassay and in the cAMP assay with GOF. The % inhibition and compound concentration showed a sigmoidal relationship, with all seven TSAb-positive sera markedly inhibited by SYD5115. An SYD5115 dose-dependent inhibition of M22 (10 ng/mL, 6 hours)-stimulated HA and/or cAMP-release from GOF was observed. Strong SYD5115-induced inhibitions of M22-stimulated cAMP production in GOF were registered with SYD5115 concentrations of 1 (p = 0.0029), 10 (p < 0.0001), 100 (p < 0.0001), 1,000 (p < 0.0001), and 10,000 (p < 0.0001) nM, respectively. SYD5115-induced inhibition of M22-stimulated HA production was noted with SYD5115 concentrations of 100 (p = 0.0392), 1000 (p = 0.0431), and 10,000 (p = 0.0245) nM, respectively. The inhibitory activity of SYD5115 was confirmed in a human osteosarcoma U2OS cell line stably expressing human TSH-R with cAMP as readout. SYD5115 induced 100% inhibition of the M22-induced cAMP levels with a potency of 193 nM. Compared with control, SYD5115 did neither impact the growth nor the migration of cultivated GOF. In addition, SYD5115 did not alter the viability of GOF.
Conclusions:
SYD5115 blocked M22- and TSAb-induced TSH-R activity with a nanomolar potency in TSH-R-overexpressed CHO cells as well as primary GOF, which demonstrates the ability of this small molecule to block TSH-R overactivity.
Introduction
Graves' hyperthyroidism (GH) and the associated orbitopathy (GO) are induced by specific thyrotropin receptor (TSH-R) autoantibodies (Ab). Orbital fibroblasts (OF) expressing the TSH-R are activated by stimulatory TSH-R-Ab (TSAb) inducing cyclic adenosine monophosphate (cAMP) increase and secretion of hyaluronic acid (HA). This activation additionally results in inflammation (1). The current treatment of GH is far from adequate. Nowadays, thionamide antithyroid drugs are the first line treatment of GH (2). However, the response rate is ∼50% only and patients may experience several dose-related adverse events (2,3). As TSH-R is expressed on orbital target cells, a novel treatment approach could both inhibit the TSH-R activity in the thyrocytes and in OF (3,4). Also, in extrathyroidal manifestations of GH, medical treatment is far from optimal. Hence, there is an unmet need for innovative and specific therapies for both GH and extrathyroidal involvement (5 –9).
TSH-R antagonizing compounds have been introduced in several preclinical in vitro studies (6 –8,10 –14). These compounds bind allosterically to the TSH-R and show varying degrees of selectivity over the closely related follicle-stimulating hormone (FSH) and luteinizing hormone (LH) receptors. Using a drug-like antagonist NCGC00229600 (11,12), the TSH- and human monoclonal antibody (mAb) M22-mediated TSH-R stimulation of cAMP was significantly reduced. A low-molecular-weight (LMW), nanomolar potent, allosteric antagonist of the TSH-R, ORG274179-0, completely inhibited the recombinant TSH, GH immunoglobulin G (IgG), and M22- induced cAMP production in differentiated OF, TSH-R Chinese-hamster-ovary (CHO) cells, and Fischer rat thyroid (FRTL-5) cells (7,8).
Most literature compounds show TSH antagonism with half inhibitory concentrations (IC50s) in the micromolar range (ca 1–20 μM), except for ORG 274179-0, which showed higher potency with IC50s in the range of 10 nM in several assays with different cellular backgrounds (7,8,10 –14). The above-cited compounds have not progressed into the clinic. However, the need for novel treatment options for GH and GO patients remains. In this study, we evaluated a novel nanomolar potent LMW TSH-R antagonist, SYD5115, in vitro in cell lines and primary Graves' OF (GOF). In the following experiments, we are demonstrating the potency of this novel small compound to inhibit TSH-R overactivity without altering the viability of primary human OF.
Materials and Methods
SYD5115
SYD5115 ((S)-3,3-dimethylbutan-2-yl (5′-acetyl-4,4-difluoro-6′,6′-dimethyl-5′,6′-dihydrospiro [cyclohexane-1,7′-pyrrolo [3,2-d]pyrimidin]-2′-yl)carbamate) was obtained as a colorless solid at Byondis, B.V., Nijmegen, The Netherlands. The molecule was identified by optimization of the properties of ORG 274179-0 by systematically varying the core structure and the appending functional groups. The optimization process, synthesis details, and physicochemical and biological characterization (in vitro and in vivo) are summarized below (15).
SYD5115 consists of an N-acetylated dihydropyrrole pyrimidine core containing several substituents such as a difluorinated-spiro cyclohexyl ring and a carbamate group on the pyrimidine ring, derived from the chiral alcohol (S)-3,3-dimethyl-2-butanol (Fig. 1). The compound has favorable pharmacochemical properties. It is soluble in aqueous media (20 mg/L at pH 7.4) and is moderately lipophilic (calcLogP = 3.9). SYD5115 has a purity of >99%, as determined by high-performance liquid chromatography (HPLC) and nuclear magnetic resonance spectroscopy analysis. The enantiomeric excess was determined to be 99.0% by chiral HPLC. The compound was used as a 10 mM (4.39 mg/mL) stock solution in dimethyl sulfoxide (DMSO) that could be stored for months at 4°C in the dark.
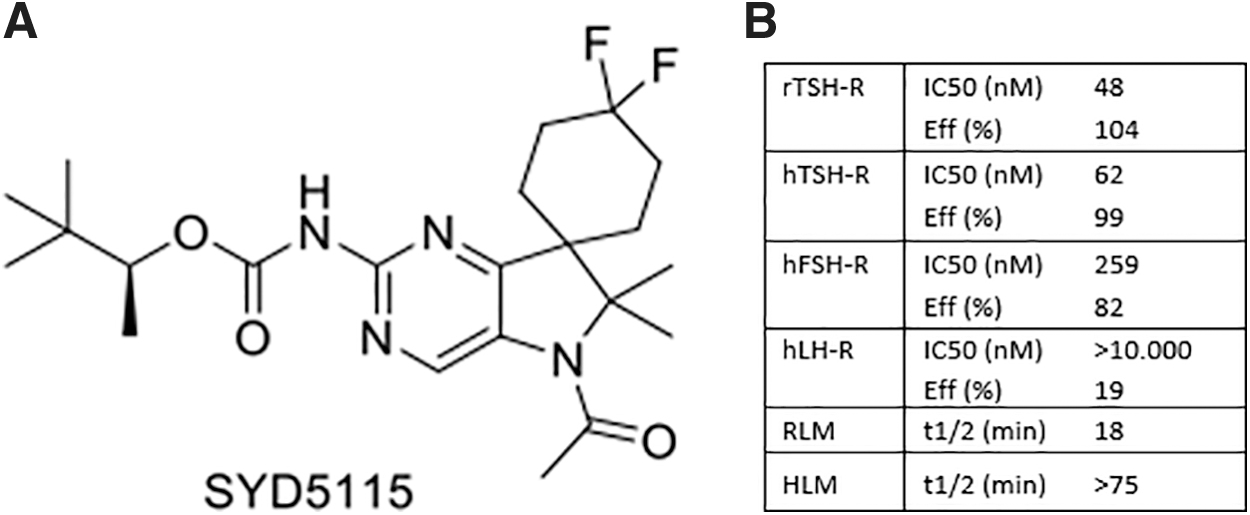
SYD5115 is a nanomolar potent LMW TSH-R antagonist with about fivefold selectivity over the human FSH receptor as measured by a cAMP response in human embryonic kidney cells (HEK293). The compound shows no significant activity, ago-, nor antagonistic effect (up to 10 μM concentration) on the LH receptor (LH-R), measured by cAMP response in HEK293 cells. SYD5115 has a moderate stability when incubated with rat liver microsomes with a half-life of 18 minutes, and a good metabolic stability in human liver microsomes with a half-life of 84 minutes, which corresponds to an intrinsic clearance (Clint) of 16 μL/min/mg protein (Fig. 1B and Supplementary Table S1). SYD5115 is expected to have medium clearance in human, although a human dose has not been predicted yet.
Human dose prediction is based on in vivo pharmacokinetic (PK) data in several species and on efficacy data in in vitro and in vivo PK/pharmacodynamic (PD) relationship. In rat, SYD5115 has a good in vivo exposure after oral dosing and favorable oral bioavailability, C max concentration, and half-life time. In a rat M22 model, SYD5115 blocked the agonistic action of a TSH-stimulating antibody after a single oral dose. The in vivo TSH-antagonistic effect of SYD5115 in rat correlated with the plasma concentration of the compound over time (15).
Culture of primary GOF
Orbital connective tissue was obtained from patients with severe GO who underwent orbital decompression surgery at the joint thyroid-eye-clinic of the JGU Medical Center, Mainz, Germany. Patients were classified with phenotypically overt and clinically severe GO, according to the Guidelines of the European Group on Graves'orbitopathy (EUGOGO) for the management of GO (5). The collection of patients' tissues and sera was approved by the Ethics Committee of the Medical Chamber of the State Rhineland-Palatinate, Germany, and by the Institutional Review Board of the JGU Medical Center. Written informed consent was received from all patients before collection of blood and tissue samples (16).
Cultures of GOF were initiated according to reference (6). In brief, tissue was minced and digested in collagenase IV solution (Gibco™, MA). Purified cells and remaining tissue pieces were plated in culture dishes. Cells were cultured in serum-free F-medium (fetal bovine serum [FBS]) in a humidified 5% CO2 incubator. All experiments were performed at passage three to limit the loss of TSH-R expression through dedifferentiation (6). For the assays, the cells were counted previously by the Luna-FL automated fluorescence cell counter (Logos Biosystems, South Korea) to sow ∼10,000 cells per well (96-well plate). After 4 days of cultivation in F-medium (37°C and 5% CO2), the cells reached confluence. Cell growth and morphology were documented daily.
Wild-type and Mc4 CHO cells
Wild-type (wt.) and chimeric TSH-R CHO (Mc4) cells were used as previously described (17 –19). Compound dilution series in reaction buffer (RB) were performed twice in wt. and Mc4 cells. The results of both runs were graphed, and a dose–response curve was drawn using GraphPad Prism version 9 (GraphPad Software, Inc., San Diego, CA). Half-maximal effective concentration (EC50) values were calculated for adequate comparison between both runs and each cell line.
Cell cultures of HEK293 cells
HEK293 cells stably expressing hTSH-R, human follicle-stimulating hormone receptor (hFSH-R), or human luteinizing hormone/choriongonadotropin receptor (hLHCG-R) (HiTSeeker cell lines; Innoprot, Derio, Spain) were grown in DMEM (Gibco) containing 10% FBS (Gibco) and supplemented with geneticin (250 μg/mL) in a humidified atmosphere in 5% CO2 at 37°C. FRTL-5 cells (ECACC) were grown in Coon's F12 modified medium (Biochrom) with
Functional cell-based TSH-R-Ab bioassays
The stimulatory (TSAb) and blocking (TBAb) reporter cell-based bioassays (Thyretain®; Quidelortho, San Diego, CA) were performed according to the manufacturer's instructions and as previously described (20,21). The blocking activity of the provided compounds was gauged utilizing the TBAb reporter bioassay, and a custom solution of M22 in Thyretain™ RB was used. Average relative light units (RLU) and coefficient of variation were calculated from each duplicate. The percent inhibition was calculated from the average RLU of the reference control and compound dilution as stated: % Inhibition = (average reference RLU − average compound dilution RLU)/(average reference RLU) × 100.
Working solution
The reporter bioassays provide a working solution containing bovine TSH (bTSH). To examine the effects of SYD5115 to antagonize the stimulating effect of TSAb, a custom working solution, consisting of M22 and RB, was used. The purely human monoclonal TSAb (M22) was purchased at RSR Ltd. (Cardiff, United Kingdom) (22). To determine the proper concentration of M22, the EC80 value was determined by creating a dilution series with M22 and RB. Concentrations of the dilution series ranged from 24 to 0.0015 ng/mL. Since the dilutions were pipetted into the wells with a further 1:11 dilution in RB, the final concentrations of M22 ranged from 7.2727 to 0.0004 ng/mL.
To ensure proper baseline stimulation of both wt. and chimeric TSH-R CHO cells, we used the twofold EC80 (1.6 ng/mL M22). The custom working solution was then used for the TBAb reporter bioassay to examine the effectiveness of SYD5115 to block M22-induced CHO stimulation.
Compound dilution series
SYD5115 was diluted in 100% DMSO. Subsequently, the compounds were diluted either with RB (Quidelortho) or serum from healthy donors (normal serum [NS]). After adding the working solution, the concentration of the respective compound on the 96 well-plate ranged from 0.32 to 1000 nM. The final DMSO concentration was 0.45%. We carried out two runs to examine reproducibility. The same applied to ORG 274179-0, which is a nanomolar potent allosteric TSH-R-antagonist (half-maximal inhibition concentration IC50 = 11 nM) (8,23).
Testing SYD5115 against TSAb-positive serum
The blocking properties of SYD5115 were tested in the TSAb bioassay (in duplicate) according to the manufacturer's instructions, using TSAb-positive sera from seven GH patients. The collected sera were then used instead of the working solution containing bTSH. SYD5115 was diluted in concentrations of 0.32 to 10,000 nM in RB.
cAMP detection in U2OS-hTSH-R cell line
For the measurement of cAMP production in the PathHunter® U2OS—hTSH-R β-Arrestin-1 Cell Line (Eurofins DiscoverX, Fremont, CA), the LANCE® Ultra cAMP Kit of PerkinElmer (Waltham, MA) was used. The TSH-R antagonist SYD5115 was diluted to the desired concentration in assay buffer, containing DMEM (Gibco) supplemented with 0.1% BSA (Sigma-Aldrich) and 20 μL rolipram (Sigma-Aldrich). Both 2.5 μL of M22 (RSR Ltd.) diluted in assay buffer and 2.5 μL of assay buffer with or without a concentration range of SYD5115 (final DMSO concentration of 1%) were added to wells of a 384-well OptiPlate™ (PerkinElmer). A 10 mM DMSO stock solution has been used for all in vitro assays, also during the optimization phase of all other analogs. Dilution to ca 1% DMSO in buffer/medium is a standard protocol in our in vitro assessment of the compounds.
The concentration range of M22 was tested in the agonistic format. The concentrations of M22 in the antagonist assays amounted to 80% (48.4 ng/mL) of those used to achieve the maximum effect on the agonist response. Then 5 μL of U2OS-hTSH-R cells (2000 cells/well) were added per well. The cells were diluted in assay buffer (DMEM supplemented with 0.1% BSA and 20 μM rolipram). The assay plates were incubated for 1 hour at 37°C in 5% CO2. The reaction was stopped by adding 5 μL of Eu-cAMP tracer working solution and 5 μL of ULight™-anti-cAMP working solution diluted in cAMP detection buffer. After 1 hour at room temperature in the dark, time-resolved fluorescence resonance energy transfer (TR-FRET) signal (fluorescent emission at 615 and 665 nm) was measured on an Envision plate-reader. The ratio was calculated by dividing fluorescence intensity at 665 nm with the fluorescence intensity at 615 nm multiplied with 10,000.
Measurement of cAMP and calculation of IC50
For the measurement of cAMP production in HiTSeeker HEK293-hTSH-R, HEK293-hFSH-R, HEK293-hLHCG-R, or FRTL-5 (rTSH-R) cell lines, the LANCE Ultra cAMP Kit of PerkinElmer was used. The TSH-antagonists were diluted to the desired concentration in assay buffer, containing DMEM (Gibco) supplemented with 0.1% BSA (Sigma-Aldrich) and 20 μL rolipram (Sigma-Aldrich). 2.5 μL of agonist (bTSH [Sigma-Aldrich], rec hFSH [Puregon], rec hLH [Luveris], or M22 [RSR Ltd.], respectively) diluted in assay buffer and 2.5 μL of assay buffer with or without the concentration range of TSH-R-antagonists (final DMSO concentration of 1%) were added to wells of a PerkinElmer 384-well OptiPlate. The concentration range of the agonists was tested in the agonistic format.
The concentrations of bTSH, rec hFSH, and rec hLH in the antagonist assays amounted to 80% (10 nM bTSH or 5 nM M22 for FRTL-5 [rTSH-R], 0.1 or 0.32 nM bTSH for hTSH-R, 50 pM rec hFSH, and 7.5 pM rec hLH, respectively) of those used to achieve the maximum effect on the agonist response. Then 5 μL of FRTL-5 or HEK293 cells stably transfected with hTSH-R, hFSH-R, or hLHCG-R (2000 cells/well) was added per well. The cells were diluted in assay buffer (DMEM supplemented with 0.1% BSA and 20 μM rolipram). The assay plates were incubated for 1 hour at 37°C in 5% CO2.
Measurement of IP-one Gq HTRF
For the measurement of Gq signaling in HEK-hTSH-R cells, the IP-one Gq HTRF® kit (Cisbio) was used. Cells (5000 c/well) were plated 1 day before the assay in 20 μL cell culture medium (DMEM +10% FBS Qualified [both Gibco]) per well of a PerkinElmer 384-well CulturePlate™ and incubated at 37°C and 5% CO2. The next day the culture medium was removed and 7 μL of assay buffer with 50 mM LiCl was added to the cells. 3.5 μL of agonist (bTSH; Sigma-Aldrich) diluted in assay buffer and 3.5 μL of assay buffer with or without the concentration range of TSH-R-antagonists (final DMSO concentration of 1%) were added to the wells. The TSH-antagonists were diluted to the desired concentration in assay buffer. The concentration range of the agonist was tested in the agonistic format.
The concentrations of bTSH in the antagonist assay amounted to 80% (400 nM bTSH for HEK-293-hTSH-R) of those used to achieve the maximum effect on the agonist response. The assay plates were incubated for 3 hours at 37°C in 5% CO2. The reaction was stopped by adding 3 μL IP1-d2 working solution and 3 μL anti-IP1-Cryptate working. After 1 hour at room temperature in the dark, TR-FRET signal (fluorescent emission at 615 and 665 nm) was measured on an Envision plate-reader. The ratio was calculated by dividing fluorescence intensity at 665 nm by the fluorescence intensity at 615 nm multiplied with 10,000.
Measurement of β-arrestin translocation
For the measurement of β-arrestin translocation in PathHunter U2OS-hTSH-R β-Arrestin-1 Cell Line (Eurofins DiscoverX), the PathHunter Detection Kit was used. Cells (3500 c/well) were plated 1 day before the assay in 20 μL AssayComplete Cell Plating reagent (PathHunter) per well of a PerkinElmer 384-well CulturePlate and incubated at 37°C and 5% CO2. The TSH-antagonists were diluted to the desired concentration in compound dilution buffer, containing PBS (Lonza) supplemented with 0.1% BSA (Sigma-Aldrich). The concentration range of TSH-R-antagonists (final DMSO concentration of 1%) was a 30-minute preincubation at room temperature in the dark before the agonist was added. 2.5 μL of agonist bTSH (Sigma-Aldrich) diluted in compound dilution buffer was added to wells. The concentration range of agonists was tested in the agonistic format.
The concentrations of bTSH in the antagonist assay amounted to 80% (100 nM bTSH for U2OS-hTSH-R β-arrestin-1) of those used to achieve the maximum effect on the agonist response. The assay plates were incubated for 6 hours at room temperature. The reaction was stopped by adding 12.5 μL of working detection solution. After 1 hour at room temperature in the dark, chemiluminescent signal was measured on an Envision plate-reader.
Measurement of HA in inhibited GOF
After 24 hours of coincubation (37°C and 5% CO2) with 10 ng/mL M22 mAb and five dilutions (10,000, 1000, 100, 10, and 1 nM) of the inhibitory antagonists SYD5115 and Org 274179-0 in RB, HA was measured with the supernatants of the GOF (n = 4). HA release was quantified (in duplicate) with an enzyme-linked immunosorbent assay (ELISA) kit (Echelon Biosciences, Salt Lake City, UT) according to the manufacturer's instructions (24).
cAMP measurement in inhibited GOF
To detect the cAMP signal, the cells were stimulated with 1.6 ng/mL M22 mAb for 30 minutes. M22 was diluted in RB, which also contained 0.75 mM 3-isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) to avoid cAMP decay. SYD5115 and Org 274179-0 were used as described above for the coincubation. Five dilutions (10,000, 1000, 100, 10, and 1 nM) were tested for SYD5115 and ORG 274170-0. The stimulation was terminated by aspiration of the medium and lysis of the GOF cells (n = 3). The lysate was taken to measure the cAMP signal by a cAMP-screen system (Applied Biosystems, Bedford, MA) according to the 96-well assay protocol of the manufacturer's reference. Measurement of cAMP release was done in duplicate.
Scratch assay
To analyze cell growth and migration under the influence of SYD5115, the scratch assay (25) was performed with human GOF. The scratch test measures the time interval for the cells to close a standardized scratch within a confluent cell layer through growth and migration. As a control, the same cells, incubated in F-medium only, were used. For reproducibility, a device consisting of two 6-well plates and magnets was used. The magnets were pulled through the confluent cell layer by moving the six-well plate, thus forming a scratch (21). The width of the scratch was evaluated using ImageJ (ImageJ software) by dividing the plate into black (confluent cell layer) and white (scratch) and comparing the percent of pixels of both colors in pictures taken over a certain course of time.
Both SYD5115 and ORG 274179-0 were used in the final concentration of 10,000 nM diluted in DMSO and F-medium. The final DMSO concentration was 0.45%.
MTT-3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide-assay
The antagonist effects on cell viability were evaluated with the MTT assay kit (Abcam, Cambridge, United Kingdom). The MTT assay is a colorimetric assay, used to measure the cellular metabolic activity as an indicator of proliferation, cell viability, and cytotoxicity. It is based on the reduction of MTT, a yellow tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] to purple formazan crystals by viable cells containing NAD(P)H-dependent oxidoreductase enzymes. The resulting colored solution is quantified by measuring absorbance at an optical density of 590 nm. The darker the solution, the greater the number of metabolically active cells. The measured absorbance is therefore proportional to the number of viable cells. All GOF strains were exposed to SYD5115 and Org 274179-0, and incubated for 48 hours. The antagonists were diluted in F-medium and the assay was performed according to the manufacturer's instructions.
Data analysis and statistical calculations
Data points of concentration–effect curves were fitted using GraphPad Prism version 9 (GraphPad Software, Inc., San Diego, CA) using nonlinear regression with curve fit method log(inhibitor) versus response (ratio) (four parameters). GraphPad Prism version 9 (GraphPad Software, Inc.) was used to perform statistical analysis. The Dunnett's multiple comparisons test was applied to compare the inhibitory effect of SYD5115 on HA and cAMP release from GOF.
Results
SYD5115 potently inhibits M22-induced TSH-R activation
To study the inhibitory activity of SYD5115 on the human stimulatory mAb (M22)-induced TSH-R activation, a luciferase-based reporter bioassay was used in CHO cell lines expressing either a wt. human TSH-R or a chimeric receptor (Mc4). Residues 262–335 of human TSH-R are replaced with a rat lutropin-choriogonadotropin receptor segment in Mc4 to improve the sensitivity and diagnostic accuracy of the bioassay. The EC80 of M22, monitored by testing a dose–response curve of M22 on CHO wt. and Mc4 cells, was 0.79 ng/mL. SYD5115 dose dependently inhibited the M22-induced TSH-R activity with a maximum efficacy of 100% on Mc4 cells, as shown in Figure 2A. Both runs with dilutions in RB and NS showed different SYD5115 EC50 values and slightly different dose–response curves.

The potency of SYD5115 in RB was higher in comparison with the dilution series in NS, which might be related to the high plasma protein binding of SYD5115. Compared with wt. CHO cell lines, the average EC50 values of SYD5115 on Mc4 CHO cells were lower than that for wt. CHO cells (35.9 nM vs. 115.7 nM) (Fig. 2B). Similar results for ORG 274179-0 are shown in Supplementary Figure S1.
SYD5115 inhibits bTSH-induced Gαq/11 signaling and translocation of β-arrestin-1
The inhibitory activity of SYD5115 was confirmed in a human osteosarcoma U2OS cell line stably expressing human TSH-R (U2OS-hTSH-R) with cAMP as readout. In this cAMP assay, M22 was used at a fixed EC80 (48.4 ng/mL) to induce TSH-R signaling and assessed in combination with the concentration range of SYD5115. SYD5115 induced 100% inhibition of the M22-induced cAMP levels with a potency of 193 nM (Fig. 3A).
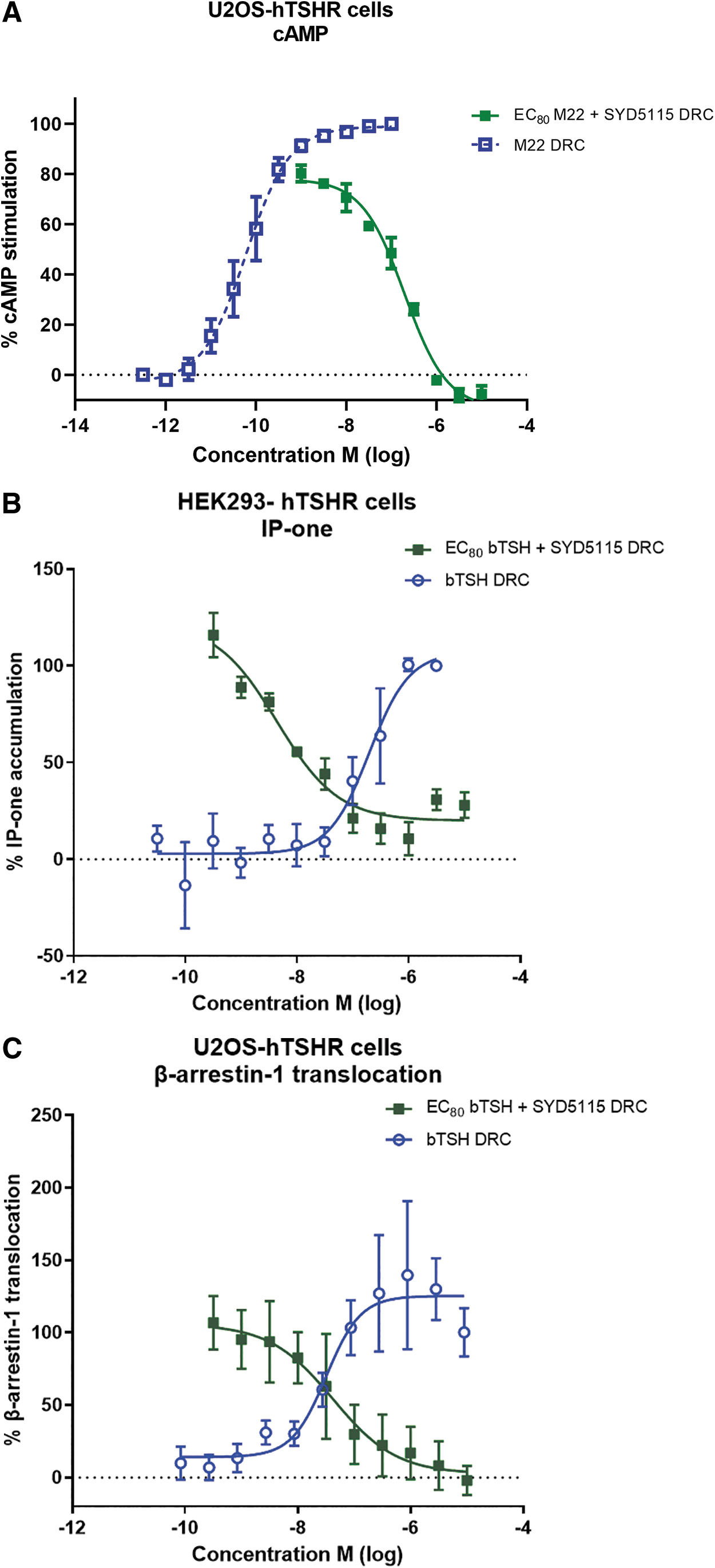
The TSH-R couples to a number of different signaling pathways via G proteins, and although the Gαs that stimulates the cAMP-protein kinase A (PKA) pathway has been considered primarily, the TSH-R can also act via Gαq/11 that stimulates phospholipase C-mediated generation of diacylglycerol and inositol-1,4,5-trisphosphate that activates the calcium and protein kinase C (PKC) pathways (26 –31). In addition, activation of TSH-R results in translocation of β-arrestin-1, which plays a role in the phosphorylation of ERK1/2, p38α, and AKT1 kinases (32,33). Next to the blocking effect of SYD5115 on the primary Gs-cAMP pathway, the effect on Gq signaling and β-arrestin-1 has been explored using an IP-one Gq HTRF kit in HEK293-hTSH-R cells and the PathHunter enzyme fragment complementation assay in the U2OS-hTSH-R cell line that stably expresses EA-β-arrestin-1.
In both assays, TSH-R activation was initiated by bTSH and a dose-dependent inhibition of SYD5115 was measured at a fixed concentration of bTSH (EC80 of bTSH 400 nM in IP-one and 100 nM in β-arrestin-1). SYD5115 blocked bTSH-induced accumulation of inositol monophosphate with an IC50 value of 4.4 nM and β-arrestin-1 recruitment with a potency of 42 nM (Fig. 3B, C).
SYD5115 blocks the stimulatory effect of TSAb-positive sera
Seven TSAb-positive sera with different levels of TSAb were used in the TBAb bioassay using chimeric Mc4 CHO cells. The % inhibition and compound concentration showed a sigmoidal relationship, with all seven TSAb-positive sera completely inhibited by SYD5115 (Fig. 4). The inhibition of the cAMP in the cell-based TSAb bioassay was similar when using two lots of the SYD5115 compound (1-year-old lot vs. a fresh one, Supplementary Fig. S2).
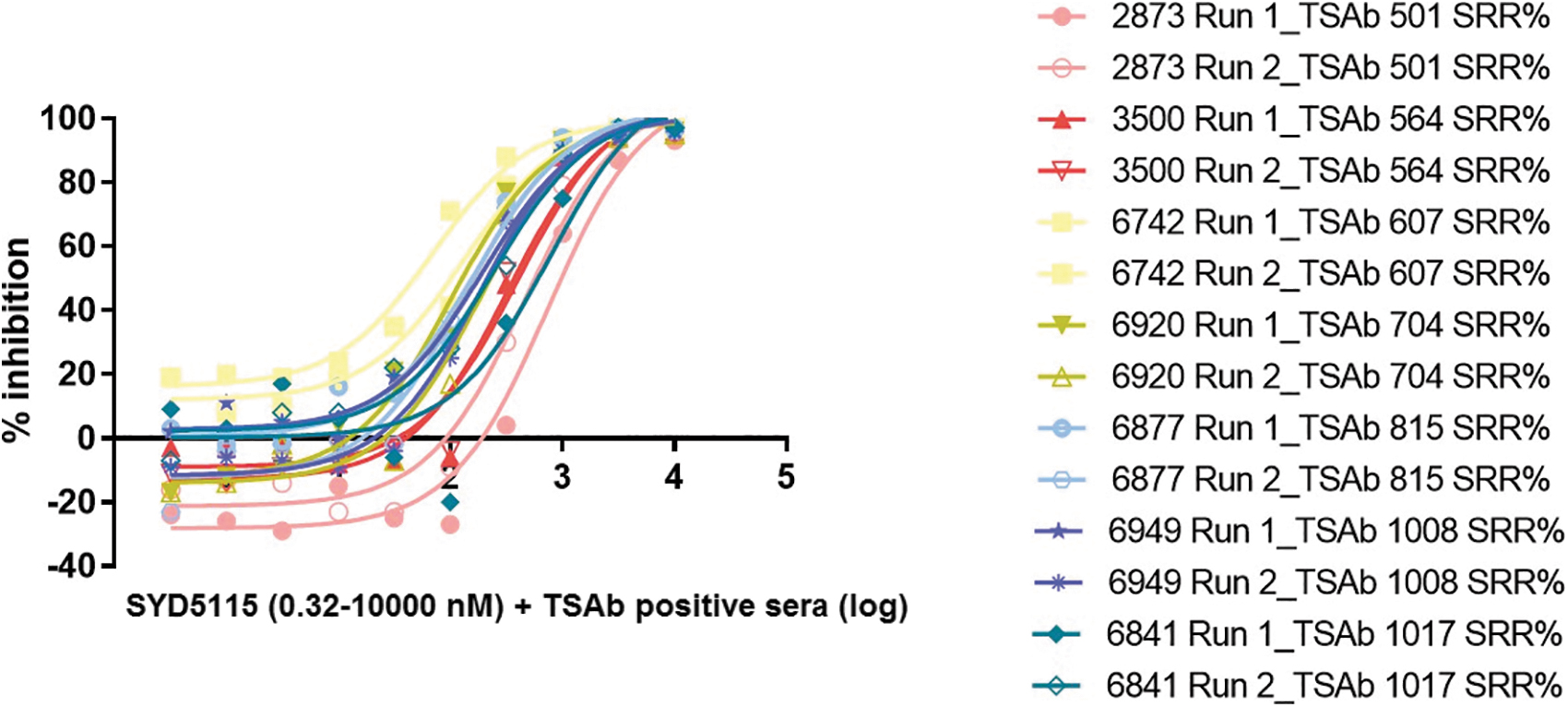
SYD5115 (0.32–10,000 nM)—Inhibition of seven different TSAb-positive sera in %, each measured in two runs (log). TSAb, TSH-R-Ab.
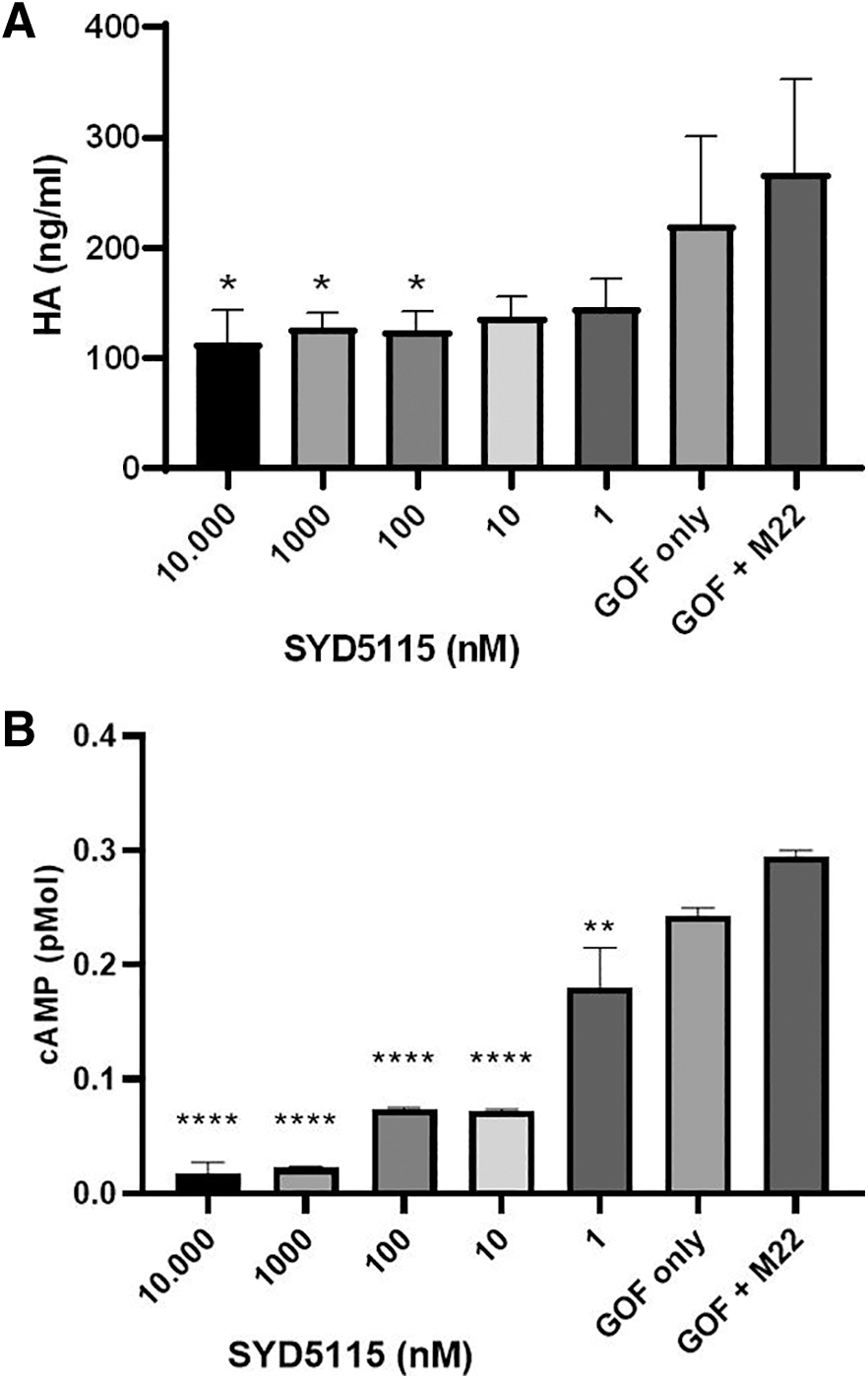
HA- and cAMP-measurements of SYD5115-inhibited GOF
SYD5115-induced inhibition of TSH-R signaling in GOF cultured cells was monitored after stimulation with M22. Five different concentrations of SYD5115 were measured after stimulation of GOF by 10 ng/mL M22 mAb for 6 hours. SYD5115 induced a dose-dependent inhibition of M22-stimulated HA production from GOF (n = 4) (Fig. 5A) and cAMP-release (Fig. 5B). In detail, inhibitions were registered with SYD5115 concentrations of 100 (p = 0.0392), 1000 (p = 0.0431), and 10,000 (p = 0.0245) nM, respectively (Dunnett's multiple comparisons test). Furthermore, strong SYD5115-induced inhibitions of M22-stimulated cAMP production from GOF (n = 3) were registered with SYD5115 concentrations of 1 (p = 0.0029), 10 (p < 0.0001), 100 (p < 0.0001), 1000 (p < 0.0001), and 10,000 (p < 0.0001) nM, respectively (Dunnett's multiple comparisons test). M22-induced cAMP levels in GOF were less effectively reduced by ORG 274179-0 (Supplementary Fig. S3).
Scratch assay
Neither SYD5115 nor Org 274179-0 has a significant influence on the growth and migration of human GOF (Fig. 6A, B). As cell growth depends mainly on the insulin-like growth factor-1 receptor, both TSH-R antagonists did not impact growth/migration of GOF. The results demonstrate the nontoxicity of SYD5115, since both growth and migration of GOF did not differ from control samples.
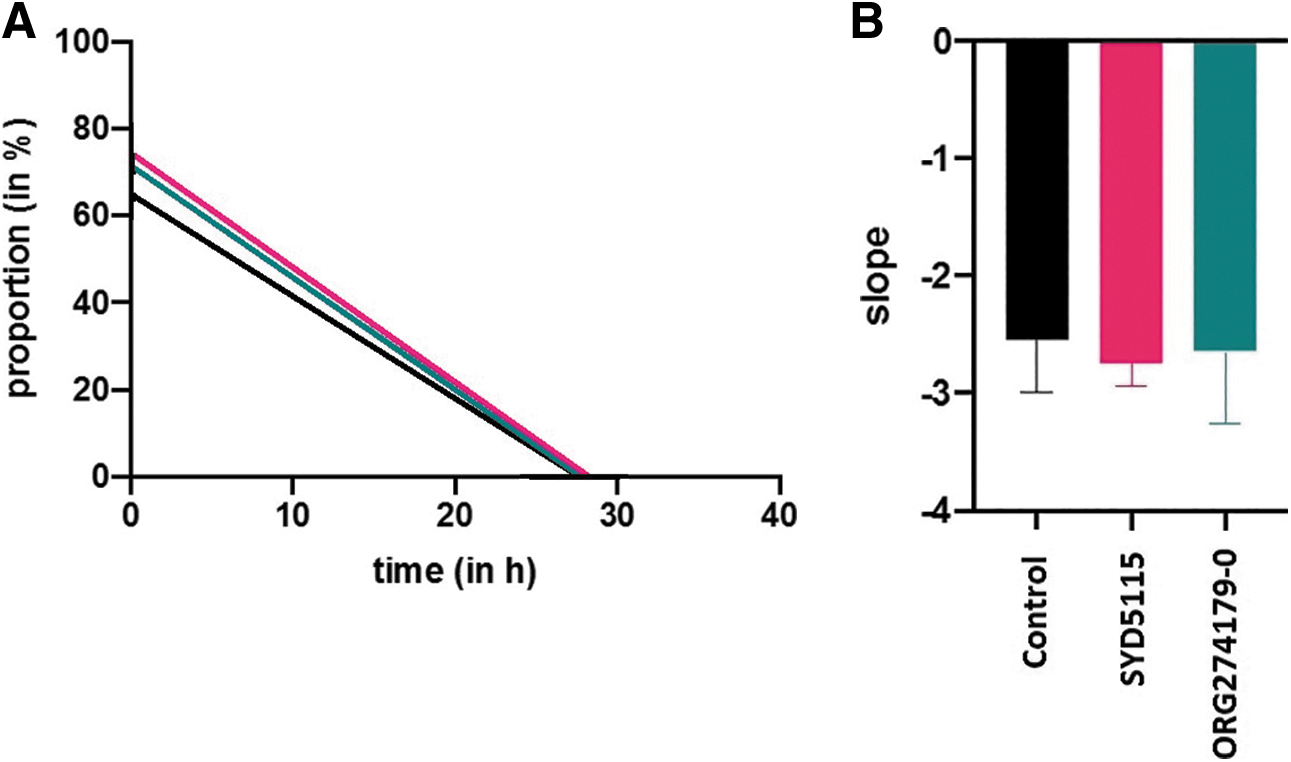
MTT-assay
SYD5115 did neither alter the viability nor the vitality of the cultivated GOF. Same results were obtained with ORG 274179-0 (Supplementary Fig. S4).
Discussion
The novel LMW compound SYD5115 effectively and dose dependently inhibited the TSH-R activity in several performed in vitro assays. In detail, this new TSH-R antagonist showed a concentration-dependent inhibition in the TSH-R-Ab blocking bioassay when being stimulated with the M22 mAb as well as being stimulated with TSAb-positive sera from several patients with GH. The compound blocked luciferase transcription in a sigmoid manner. This inhibition could be demonstrated both in genetically modified and in primary human cell cultures from GOF. We tested SYD5115 against serum samples from seven different GH patients with high to very high TSH-R-Ab concentrations (specimen-to-reference ratio [SRR%] 501-1017).
In this study, SYD5115 inhibited TSAb-mediated stimulation from all seven samples with almost 100% inhibition at 1000 nM. Furthermore, SYD5115 was able to block the cAMP increase in the primary GOF cultures. The inhibitory effect was also observed in U2OS-hTSH-R cells, as SYD5115 was able to fully block M22-induced cAMP synthesis. Similar to the results of the TSH-R-Ab blocking bioassay, cAMP production was blocked in a sigmoidal manner. Finally, the scratch and MTT assay results showed no impact on GOF cell viability, cell growth, and cell toxicity.
To test the suitability of SYD5115 as a potential new treatment, we consequently examined its ability to block the TSH-R. In GH or GO, thyrocytes or OF are overstimulated with TSH-R-Ab. These autoantibodies cause a cAMP-mediated molecular cascade, which leads to cell activation, proliferation, and secretion of hydrophilic, water-binding glycosaminoglycans, mostly HA. This leads to the well-known clinical signs and symptoms of GO. We utilized this cascade in our cAMP and HA assays, by gauging cAMP and HA concentration. In our bioassay, the intracellular increase in cAMP induces the transcription of luciferase, an enzyme emitting light signals by conversion of its substrate. The enzyme activity is measured by a luminometer and is proportional to TSH-R activation. To measure an inhibitory effect, a baseline stimulation with a standardized solution of M22 must be implemented. The TSH-R antagonistic property of the compounds can then be quantified by the reduction of the baseline stimulation.
We conducted the blocking TBAb bioassay with RB and NS, intending to create a more physiological environment. M22 was used to create baseline stimulation in the bioassays since M22 stimulates TSH-R at the extracellular domain similarly to TSH (22). In comparison with NS, we reached lower IC50 and total RLU counts with RB. We attribute this difference to polyethylene glycol (PEG), a chemical present in RB with augmentative effects on stimulating Ab binding to TSH-R or by proteins, such as albumin or plasma proteins, in NS binding to the small molecule (34). In the absence of PEG in NS, lower compound concentrations (0.32 to 31.6 nM) showed negative % inhibition in both runs. This effect was confirmed with the second run and is in line with previous research, indicating a higher affinity of stimulating Ab toward TSH-R.
In our bioassay, we observed higher RLU counts and higher sensitivity with the chimeric CHO cell line in comparison with wt. cells. The Mc4 cell line showed an eightfold higher sensitivity to ORG 274179-0 and a fourfold higher sensitivity to SYD511 in comparison with wt. cells. This difference in outcome has been previously reported and is explained by the chimeric composition of the TSH-R in the Mc4 cell line (23,35).
Over the years, several LMW antagonists or inverse agonists (7,10,12 –14,36) affecting the TSH-R have been investigated and a few showed promising results in receptor inhibition or even inhibition of basal signaling. Van Zeijl et al. examined ORG 274179-0 used in this study as a direct comparison. Neumann et al. also tested several inverse agonists and neutral antagonists (10,11). The advantage of inverse agonists is that they are additionally able to block basal cell activity. Neumann et al. further examined a difference when the cells were stimulated to differentiate into preadipocyte cell cultures, before using them in an assay. Differentiated cultures mimic a later stage of disease (12).
To date none of these LMW compounds made it to clinical testing. This could be due to the lack of potency, in vivo exposure and/or efficacy, toxicity, or lack of selectivity over the closely related FSH and/or LH receptors. In previous research, ORG 274179-0 was able to block signaling in wt. cells as well as in cells with autonomous functioning TSH-R mutants. Our results are in line with the above research findings, revealing the inhibitory effect of SYD5115 in wt. cells. However, it is still not known if SYD5115 is able to decrease the signaling of autonomous TSH-R mutants. If this is the case, SYD5115 could be effective in treating autonomously functioning thyroid adenomas (37).
The authors acknowledge the following limitations of their research: (1) testing the new compound with several cell lines instead of evaluating its potency with human thyroid cells in primary cultures, (2) the experiments with GOF were performed only with stimulation with M22 and not with TSAb-positive sera, (3) no modeling of the molecule binding was provided.
In conclusion, SYD5115 in vitro potency on the TSH-R shown in cell lines and primary GOF as described here appeared praiseful and as such does show that an LMW approach to inhibit TSH-R activity might turn out to be successful in the future.
Footnotes
Acknowledgment
The authors thank Dr. Tanja Diana, JGU Thyroid Lab, for data collection and fruitful discussions.
Authors' Contributions
G.J.K.: Conceptualization, funding acquisition, methodology, resources, supervision, project administration, validation, visualization, writing—original draft, and writing—review and editing. A.G.: Investigation, methodology, formal analysis, software, validation, visualization, and writing—original draft. L.S.: Methodology, investigation, formal analysis, validation, visualization, and writing—review and editing. T.W.: Methodology, investigation, visualization, and writing. J.H.: Methodology, investigation, formal analysis, and writing. L.F.: Methodology, formal analysis, review, and editing. J.W.: Data curation, methodology, formal analysis, visualization, and writing—review and editing. M.M.C.v.d.L.: Conceptualization, resources, methodology, formal analysis, and writing—review and editing. T.A.E.v.A.: Conceptualization, resources, methodology, formal analysis, and writing—review and editing. R.J.A.: Conceptualization, resources, methodology, formal analysis, and writing—review and editing. W.F.J.K.: Conceptualization, resources, methodology, formal analysis, and writing—review and editing.
Ethics
Written informed consent was obtained from all participants, and the study was carried out in accordance with the Johannes Gutenberg-University (JGU) Ethics Committee and the ethical guidelines of the Helsinki Declaration. All animal experiments were performed in compliance with the European directives (2010/63/EU) and approved by the local ethics committees. Animals were housed and handled in accordance with good animal practice as defined by FELASA, in AAALAC-accredited animal facilities.
Author Disclosure Statement
G.J.K. consults for Quidelortho, San Diego, CA. M.M.C.v.d.L. is employee of Byondis, NL. T.A.E.v.A. is employee of Byondis, NL. R.J.A. is employee of Byondis, NL. W.F.J.K. is employee of Byondis, NL. The other authors have nothing to disclose.
Funding Information
The JGU Medical Center received research-associated funding from Byondis B.V., Nijmegen, The Netherlands, and from Quidelortho, USA.
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4