
Abstract
Background:
Very little was known about the molecular pathogenesis of thyroid cancer until the late 1980s. As part of the Centennial celebration of the American Thyroid Association, we review the historical discoveries that contributed to our current understanding of the genetic underpinnings of thyroid cancer.
Summary:
The pace of discovery was heavily dependent on scientific breakthroughs in nucleic acid sequencing technology, cancer biology, thyroid development, thyroid cell signaling, and growth regulation. Accordingly, we attempt to link the primary observations on thyroid cancer molecular genetics with the methodological and scientific advances that made them possible.
Conclusions:
The major genetic drivers of the common forms of thyroid cancer are now quite well established and contribute to a significant extent to how we diagnose and treat the disease. However, many challenges remain. Future work will need to unravel the complexity of thyroid cancer ecosystems, which is likely to be a major determinant of their biological behavior and on how they respond to therapy.
Introduction
The objective of this review was to chart the history of the major discoveries leading to our current understanding of the genomic evolution of thyroid cancer and to place this information in the context of the mechanistic and technological breakthroughs that made these discoveries possible. Due to the limitations of the format of the article, we are unable to credit the work of all investigators who contributed to this field, and for this, we apologize to those who have been overlooked.
Clonal Composition of Thyroid Neoplasms
Although it may seem self-evident to current readers, until the late 1980s, it was not clear whether all thyroid tumors were clonal and thus derived from a single cell transformed through a mutation that conferred it with a fitness advantage. This was certainly the case for nodules arising within multinodular goiters (MNG) that were considered at the time to be mostly hyperplastic, and therefore at lower risk for malignancy. The technology that allowed this to be clarified arose from work by Philip Fialkow in hematological malignancies. Dr. Fialkow was a geneticist who showed that leukemias were clonal in women who were heterozygous for a variant in the X chromosome-linked G6PD gene, which resulted in G6PD proteins with distinct electrophoretic mobility (1).
Since one of the two X chromosomes is randomly inactivated in cells through DNA methylation (2), normal tissues in women heterozygous for the G6PD variant would contain a mixture of cells randomly expressing either the paternal- or maternal-derived copy of the gene, resulting in both G6PD proteins to be present in equal proportion in tissue extracts, whereas if a tumor had arisen from a single cell, all tumor cells would produce the same G6PD protein. Vogelstein refined the power of this approach by using restriction DNA enzymes that differentially digested X chromosome alleles based on their methylation status, thus enabling distinction of polyclonal from clonal cell populations by Southern blotting of DNA (3).
With this technology, several groups showed that thyroid adenomas and carcinomas were clonal and that clonal tumors also arose with some frequency within MNG (4 –6). This strategy to investigate clonality is confounded in the thyroid context by evidence that normal thyroid epithelium is organized into large stem-cell derived monoclonal patches, such that a variable subset of normal thyroid tissue extracts showed monoclonal patterns by X-chromosome inactivation analysis (6 –8), requiring tumors from these patients to be excluded from the analysis.
Discovery of Thyroid Cancer Genes Through the NIH-3T3 Cell Focus Assay
The search for genes involved in thyroid cancer pathogenesis initially relied on a method that leveraged the ability of tumor viruses to transform mouse fibroblast cell lines in vitro (9). Since many human oncogenes are structurally related to these viruses, a method was devised to identify human cancer genes based on the ability of shredded human tumor DNA to transform mouse NIH-3T3 cells, through sequential rounds of DNA isolation of transformants containing human DNA sequences and re-transfection into NIH-3T3 cells (10). Transformed fibroblast cells could be identified and isolated under the microscope because they were no longer inhibited by cell contact and tended to grow on top of each other and form little nodules on the culture dish.
Using this approach, Takahashi isolated human DNA encoding the C-terminal region of REcombined during Transfection (RET), which encompasses its kinase domain, from NIH-3T3 cells transfected with human lymphoma DNA, which was fused to an unrelated human DNA fragment. Since the RET rearrangement was not present in normal cells or in the original lymphoma DNA, he concluded that the two genes had REcombined during Transfection (hence the term RET) (11). In 1989, two Italian groups identified bona fide rearrangements of RET in papillary thyroid cancers (PTCs) using the NIH-3T3 cell focus assay (12,13) and also identified NTRK1 fusions as PTC oncogenes (13). The groups of Suarez and Lemoine also employed the NIH-3T3 cell focus assay to identify RAS mutations in thyroid cancer (14,15). Hence, three common drivers of differentiated thyroid cancers were first identified through this screening method.
The Mostly Futile Search for Copy Number Alterations in PTC
The search for tumor suppressor genes was primarily performed by screening genomic DNA for regions of loss of heterozygosity (LOH), based on the evidence that genetic lesions of both copies of these genes were required to inactivate their function, one of which often took place through copy number loss (16). By using polymorphic DNA markers, regions of recurrent copy number loss could then point out to the location of a tumor suppressor, as performed, for instance, for the identification of RB1 in retinoblastoma (17), and APC, TP53, and DCC in colorectal cancers (18). This approach was not revealing in the early studies of thyroid cancer, mostly because they primarily focused on small series of PTC, a thyroid cancer type that we now know to be diploid in about 75% of cases, and associated with infrequent recurrent copy number abnormalities (19).
Search for Genetic Drivers Based on Knowledge of Thyroid Cell Physiology
A prior understanding of the mechanisms of thyrotropin (TSH)-induced cell signaling and growth rendered the key nodes in the TSH receptor (TSHR) signaling pathway as attractive candidate oncoproteins for thyroid neoplasms. Shortly after the Nobel prize winning discovery by Sutherland of cAMP as a “second messenger” (20), TSH and other hormones were found to signal through a cAMP-dependent pathway (21). The TSHR complementary DNA (cDNA) was cloned in 1989 and found to be a member of the G protein-coupled receptor family (22). In 1990, activating somatic mutations of GNAS, the gene encoding Gsα, a GTPase that signals to activate cAMP generation, were discovered in pituitary somatotroph tumors and autonomously functioning thyroid adenomas (AFTA) (23,24).
Activating mutations of TSHR were subsequently found to be the most common driver of AFTA (25,26). Mutations of GNAS and TSHR are rare in thyroid cancer, consistent with the evidence that AFTA have a low risk of malignant transformation (19,27). Recently, a recurrent hot spot mutation (c.1712A>G;
Polymerase Chain Reaction-Based Screens for Candidate Genes
Rearrangements of RET and RAS point mutations were subsequently confirmed by several groups primarily using a candidate gene, polymerase chain reaction (PCR)-based technology (29,30), which, together with Sanger sequencing, greatly accelerated the pace of cancer gene discovery. In the early 1990s, TP53 mutations as hallmarks of anaplastic thyroid cancer (ATC) were reported by several groups by using targeted PCR-based approaches that surveyed specific hot spot exons of the gene, based on data reported in other cancer types (31 –33).
In some studies, the PCR products were sequenced directly, and in others, they underwent a preliminary screening strategy using RNase protection assays, which can identify base substitutions by hybridizing PCR-amplified single-strand DNA to a radiolabeled RNA probe encoding the wild-type sequence. DNA-RNA mismatches are detected by cleavage of the unhybridized single-strand RNA with RNAse 1, which allows triage of samples for Sanger DNA sequencing. These screening approaches could be scaled up to evaluate multiple samples on a single gel, since at the time, Sanger DNA sequencing gels were read manually, which was a slow cumbersome process. TP53 mutations were the first genetic lesion present in anaplastic but not in differentiated thyroid cancers, which began to delineate events involved in tumor microevolution.
In 1993, a major breakthrough was achieved in the understanding of the genetic basis of familial and sporadic forms of medullary thyroid carcinomas (MTC). A study by Mulligan et al. identified germline-activating mutations of the RET gene, previously found to be activated by rearrangement in PTC, to be responsible for multiple endocrine neoplasia (MEN) type 2A (MEN2A), a syndrome with MTC as its main clinical manifestation (34).
Specifically, sequencing of RET, located within a 480-kb region on chromosome 10q11.2 to which the putative MEN2A gene had been previously localized by genetic and physical mapping techniques, revealed missense point mutations and small in-frame insertions or deletions in the conserved cysteine residues at the boundary of the RET extracellular and transmembrane domains in the tumor and blood samples from MEN 2A families (34). Soon thereafter, activating mutations in the intracellular tyrosine kinase domain of RET, typically at M918T, were identified in patients with MEN2B and sporadic MTC (35,36). Discovery of RET mutations opened a new era for managing patients with familial MTC, enabling early diagnosis and guiding prophylactic thyroid surgeries for the affected family members (37).
In the year 2000, the PAX8-PPARG fusion was identified as a common genetic alteration in follicular thyroid carcinomas (FTC), the second most common type of thyroid cancer after PTC (38). The fusion was identified by mapping the genes located at the breakpoints of the recurrent t(2;3) (q13;p25) chromosomal rearrangement, which had been previously detected by cytogenetic analysis in several FTC. Soon thereafter, it became apparent that PAX8-PPARG fusions do not co-occur with RAS mutations, another very common driver in these tumors, with each of these drivers being responsible for the development of ∼40% of FTC (39). This fusion was also found in a proportion of follicular variant PTC and follicular adenomas, providing the evidence that these tumors were biologically related to FTC (40).
2003 was a particularly significant year for thyroid cancer genetics due to the identification of BRAF mutations, primarily BRAFV600E , as the most common driver event in PTC, as well as in advanced thyroid cancers (41 –44). A year earlier, BRAF had been characterized as an oncogene in melanomas, colorectal, and ovarian cancers (45). The gene codes for a cytoplasmic serine/threonine kinase that signals along the mitogen-activated protein kinase (MAPK) pathway. The V600E mutation, initially annotated as V599E, was found to lead to the strongest phosphorylation of downstream ERK1/2 compared with the much less common mutations found in the residues of the activation segment of the protein surrounding residue 600 or in the P loop (45). In thyroid cancer, BRAFV600E was found in 29–69% of PTC (41 –44), and it quickly became apparent that this mutation was restricted to PTC and poorly differentiated thyroid carcinoma (PDTC) and ATC arising from pre-existing papillary carcinoma (46).
From the first reports, it was evident that BRAF mutations did not overlap with RAS mutations or RET/PTC rearrangements in PTC, establishing that activation of the MAPK pathway by any of its effectors is sufficient for driving the development of PTC (41,44). Subsequently, genetic alterations in different nodes of the MAPK pathway were found to confer tumors with distinct PTC phenotypes. Whereas PTC carrying BRAFV600E or RET/PTC fusions typically had prominent papillary growth characteristics of classical type PTC, RAS-driven tumors displayed almost exclusively a follicular growth pattern with abundant colloid, characteristic of the follicular variant PTC (47). These discoveries increased the proportion of thyroid cancers with known genetic drivers and established the central role of MAPK activation in thyroid cancer initiation, thus laying the foundation for the development of genetic panels for thyroid cancer diagnosis and oncoprotein-targeted therapies that blossomed during the ensuing decades.
In 2013, two back-to-back articles in Science reported recurrent somatic mutations at specific nucleotides in the promoter for TERT in patients with melanomas (48,49). These mutations were also present in the germline in a rare familial form of melanoma (48). The TERT gene encodes for the telomerase reverse transcriptase, a component of the telomerase complex that prevents telomere shortening during cell division, a key process enabling cell immortalization in patients with cancer. The TERT promoter mutations in melanoma generate de novo consensus DNA binding sites for the ETS family of transcription factors, which mediate in part the transcriptional program activated by the MAPK signaling pathway, which in melanomas is driven by activating mutations of BRAF or RAS.
Since these same driver mutations are highly prevalent in thyroid cancers, several groups quickly found that TERT promoter mutations were also present in thyroid cancers, primarily in more advanced forms of differentiated thyroid cancer, as well as PDTC and ATC (50 –53). To the best of our knowledge, this was the first highly recurrent somatic mutation affecting a non-coding sequence in cancer. Moreover, it was later shown to carry important prognostic implications in patients with differentiated thyroid cancer (54). This has been demonstrated most clearly in the context of coexisting BRAFV600E mutation (55), although TERT promoter mutations are also enriched in advanced cancers driven by oncogenic fusions and mutations of RAS (56).
Activation of the phosphatidylinositol 3-kinase (PI3K) pathway is also implicated in thyroid carcinogenesis, which occurs mostly via inactivating mutations or deletions of the PTEN tumor suppressor gene, which encodes for a dual-specificity phosphatase for phosphatidylinositol (3,4,5)-trisphosphate, as well as activating mutations and copy gains of PIK3CA, the catalytic subunit of phosphoinositide 3-kinase, and activating mutations of the phosphatidylinositol (3,4,5)-trisphosphate substrate AKT1 (57 –59). Germline inactivating mutations of PTEN predispose to development of MNG, follicular adenomas, and differentiated thyroid cancers (60,61). Somatic mutations of these three genes are enriched in advanced differentiated thyroid cancers, including radioiodine-refractory cancers (58), as well as in PDTC and ATC, where they often coexist with mutations of BRAF or RAS (56,58,62).
Gene Expression Studies in Differentiated Thyroid Cancer
In addition to the expansion of knowledge on driver alterations occurring in genomic DNA of cancer cells, the broad availability of microarray technologies in the late 1990s and early 2000s enabled exploration of global gene expression changes in the disease. High-density cDNA microarrays allowed quantification of messenger RNA (mRNA) transcripts for each of thousands of genes by hybridizing tumor cDNA with gene-specific oligonucleotide probes spotted on a solid surface. The first studies using cDNA microarrays demonstrated profound changes in gene expression profiles of thyroid cancer compared with normal thyroid tissue and follicular adenomas, with consistent patterns of mRNA expression changes among PTC or FTC analyzed in the same study (63,64). However, further reports demonstrated significant differences in gene expression profiles between specific histological variants of PTC, such as classical papillary and follicular variants (65,66), and between PTC driven by BRAFV600E , RAS mutations, and RET/PTC fusions (67,68).
In the early 2000s, microRNAs (miRNAs) were recognized as a distinct class of small non-coding RNA molecules affecting post-transcriptional regulation of gene expression in different human diseases including cancer (69). In 2005, He et al. provided the first demonstration of upregulation of specific miRNAs in PTC compared with normal thyroid tissue (70). Soon thereafter, upregulation and downregulation of specific miRNAs were found in other types of differentiated and dedifferentiated thyroid cancers (71,72). Similar to mRNA profiles, significant differences in expression of individual miRNA were observed in PTC driven by BRAFV600E , RET/PTC, and RAS mutations (73).
Advent of Next-Generation Sequencing Technologies: The Cancer Genome Atlas Study of PTC
Sanger sequencing technology was the primary method for DNA sequencing for more than 30 years following its original discovery in 1977 (74). Its throughput was markedly improved following the introduction of capillary electrophoresis (CE)-based sequencing instruments in 1987, which became the primary tools used for the completion of the Human Genome Projects (75). However, it was not until the advent of massively parallel next-generation sequencing (NGS) technologies that the field of cancer genomics was truly revolutionized, by enabling large-scale studies of whole exome, whole genome, and RNA sequencing of tumor samples. The Cancer Genome Atlas (TCGA) program, a collaborative effort of the United States National Cancer Institute with the National Human Genome Research institute, was launched in 2006 and played a central role in shepherding landmark comprehensive genomic, epigenomic and transcriptomic analyses of the major types of human cancer using NGS.
TCGA elected to focus on in-depth studies of the most common type of cancer of each main cell lineage, which in the case of thyroid follicular cells was PTC. This large team science effort included a group of disease experts, some of whom unsuccessfully advocated for inclusion of the more aggressive types of thyroid cancer in the analysis because the major drivers of PTC were already known, which was not the case for PDTC and ATC. Ultimately, the decision to focus solely on the most common disease entity in our view proved to be far-sighted, as the homogeneity of the cohort allowed for robust integrative analyses of multidimensional molecular data, a rich trove of information that was published in 2014 and that to this day continues to be mined by the research community to fuel new discoveries (19). TCGA PTCs proved to be mostly diploid, to have a very low mutation burden, and were confirmed to be driven mostly by clonal nonoverlapping mutations of BRAF, RAS, and fusions of RET and NTRK.
However, whole exome, and in a smaller subset, whole-genome sequencing identified new thyroid cancer genes, among others EIF1AX, CHEK2 and PPM1D, as well as a more expanded repertoire of fusion oncoproteins (19). RAS mutants and BRAFV600E activate the MAPK pathway, but they do so with different intensity. Oncogenic RAS constitutively signals through RAF dimers, which are subject to negative feedback by ERK, resulting in a relative attenuation of the MAPK pathway flux. By contrast, BRAFV600E signals as a monomer and is partially unresponsive to negative feedback by ERK, resulting in a higher MAPK signaling output (76) (Fig. 1). Accordingly, BRAF-mutant PTCs are transcriptionally distinct from their RAS-mutant counterparts.
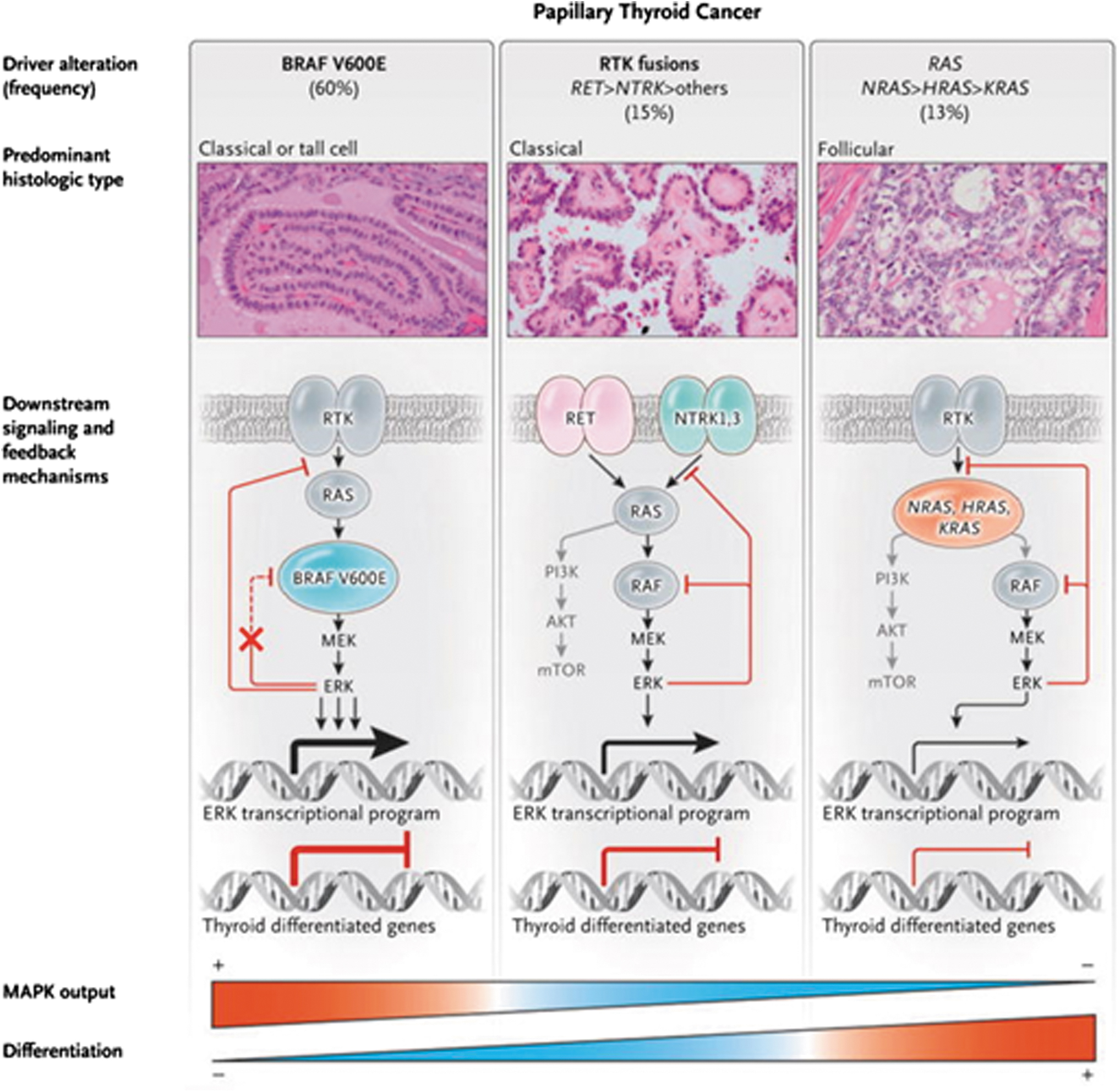
Functional consequences of driver mutations in PTCs: RTK fusions, RAS mutants, and BRAFV600E activate the MAPK pathway, but they do so with different intensity. RTK fusions such as RET/PTC and ETV6-NTRK3 and mutant RAS proteins signal through RAF dimers, which are subject to negative feedback by ERK, resulting in a constitutively active but dampened flux through the MAPK pathway. By contrast, BRAFV600E signals as a monomer and is partially unresponsive to negative feedback by ERK, resulting in a higher MAPK signaling output. The expression of genes required for thyroid cell differentiation and thyroid hormone biosynthesis is inversely correlated with the transcriptional output of the MAPK pathway. MAPK, mitogen-activated protein kinase; PTC, papillary thyroid cancer; RET, REcombined during Transfection; RTK, receptor tyrosine kinase. From N Engl J Med, Fagin JA, Wells SA Jr., Biological and Clinical Perspectives on Thyroid Cancer, v375:1054–1067. Copyright © (2016) Massachusetts Medical Society. Reprinted with permission.
The integration of the driver mutations with the transcriptomic data from RNA sequencing allowed the derivation of a transcriptomic score, termed BRAF-RAS score (BRS), which categorized individual tumors based on their gene expression alignment with either a BRAFV600E or an RAS-mutant tumor. The BRS associates strongly with PTC pathological features, metastatic tropism, and differentiation state (Fig. 2). BRAF-like PTCs are typically classic or tall cell variants of PTC, more likely to show infiltrative border, metastasize to lymph nodes before spreading to distant sites, and be relatively refractory to radioactive iodine (RAI).
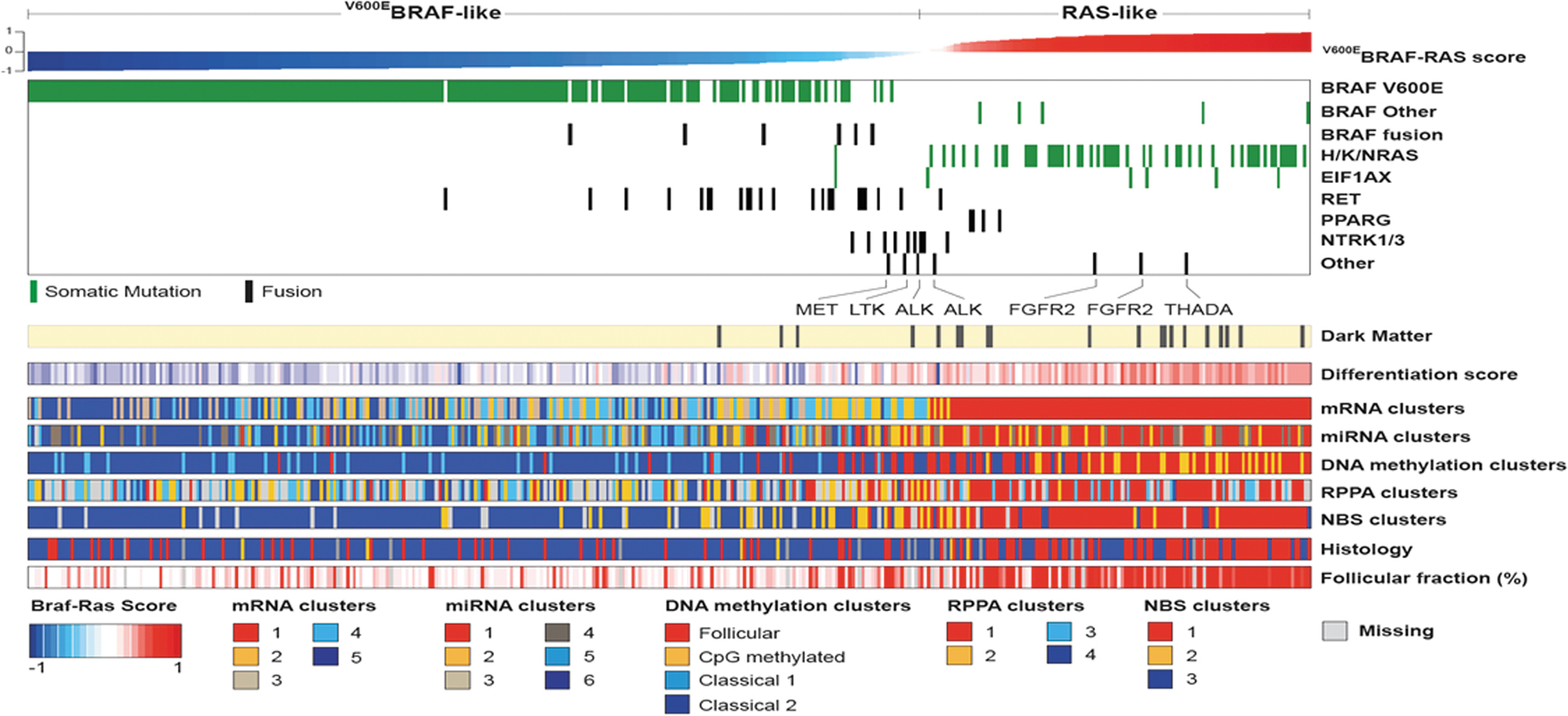
BRAF-like and RAS-like papillary carcinomas: The transcriptomic, microRNA, and methylome profiles of BRAFV600E-mutant and RAS-mutant PTC are distinct. A 71-gene signature, termed the BRS, was generated by TCGA to classify tumors as either BRAF-like or RAS-like. PTCs harboring other drivers can be categorized based on this signature: for example, RET fusions tend to be BRAF-like, whereas NTRK fusions lie somewhere in between. The BRS correlates with the histological characteristics of the tumors, their differentiation state, as well as their metastatic tropism. BRS, BRAF-RAS score; TCGA, The Cancer Genome Atlas. Reprinted with permission from Cell 2014;159:676–690 (ref. 19).
By contrast, RAS-like PTCs are follicular variants, tend to be encapsulated, can invade blood vessels and metastasize hematogenously bypassing regional lymph nodes, and are more RAI-avid. These concepts are now beginning to be incorporated into clinical practice. Other TCGA molecular platforms, such as miRNA sequencing, DNA methylation profiling, and proteomics, were also highly revealing and collectively have resulted in a deeper and more fundamental understanding of the biology of these common cancers.
Genetics of Oncocytic (Hurthle Cell) Tumors
Until recently, the molecular pathogenesis of oncocytic (Hurthle) cell tumors, composed of cells with innumerable abnormal mitochondria, remained poorly understood. Although loss-of-function mitochondrial DNA mutations in complex I genes had been commonly observed in these cells (77,78), most of these tumors lacked the nuclear DNA drivers seen in other types of thyroid cancer. In 2012, Corver et al. reported that most oncocytic carcinomas had a highly unusual pattern of chromosomal copy number alterations called genome near-haploidization (79). It involved either the loss of one of the two copies of most chromosomes resulting in monosomy or alternatively uniparental disomy, in which the remaining chromosome was duplicated so that the copies were both either paternal or maternally-derived. Such changes affected multiple chromosomes but spared chromosome 7, which always retained heterozygosity (79).
In 2018, two back-to-back publications provided comprehensive genomic characterization of oncocytic carcinomas using whole exome and whole transcriptome sequencing among other techniques (80,81). They showed that these tumors were characterized by (i) recurrent homoplasmic mutations of mitochondrial genes, primarily those encoding complex I enzymes of the electron transport chain; (ii) widespread chromosomal copy number alterations resulting in genome near-haploidization (which was found in most of these tumors); and (iii) nuclear DNA mutations in a small proportion of these cancers, which included RAS mutations, typically seen in diploid tumors, as well as TERT and TP53 mutations, seen in tumors with and without chromosomal copy number alterations (80,81) (Fig. 3).
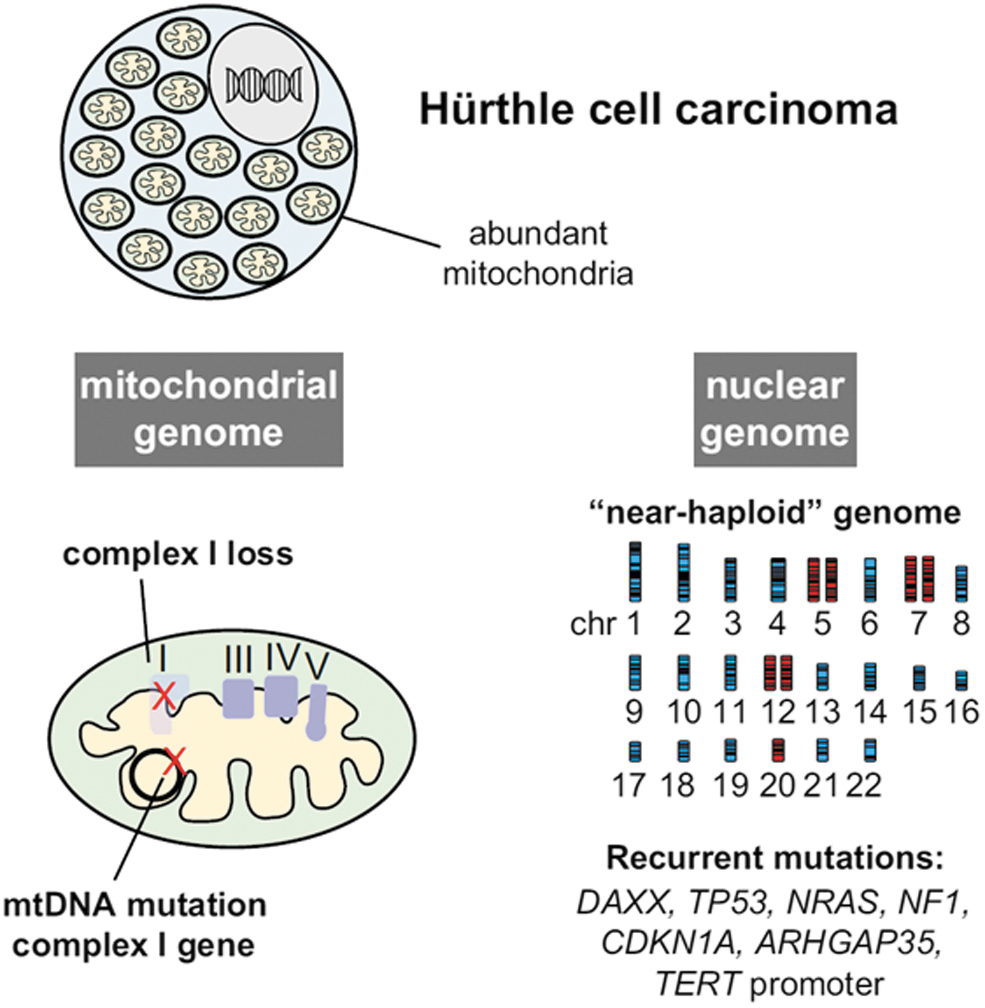
Genetics of Hurthle cell carcinomas: Hurthle cell (oncocytic) tumors are driven by alterations of the nuclear and mitochondrial genomes. Mutations of mtDNA primarily involve loss-of-function complex I gene mutations. Most of these cancers also develop widespread loss of one of the two chromosome alleles leading to a near-haploid genome. Nuclear DNA mutations are present in a smaller fraction of Hurthle cell cancers, with TP53 and TERT mutations found more commonly in widely invasive cancers. mtDNA, mitochondrial DNA. Reprinted with permission from Cancer Cell 2018;34:242.e5–255.e5 (ref. 81).
Genome near-haploidization was subsequently noted not to be restricted to oncocytic carcinomas, as it is present in about one third of oncocytic adenomas, which likely serve as precursor lesions for oncocytic carcinomas (82). The unique genetic mechanisms of oncocytic tumor development provided strong support for reclassification of these tumors as an independent pathologic entity and not, as previously thought, as variants of FTC and adenoma.
Cancer Exome Sequencing Panels Propel Discovery of Genetic Drivers of PDTC and ATC
PDTC and ATC are uncommon types of thyroid cancer, yet they are of paramount interest because of their association with poor clinical outcomes. So far neither TCGA nor other large cancer genome sequencing consortia have focused on advanced thyroid cancers. An early whole-genome sequencing study of 22 ATC identified MAPK pathways drivers as common recurrent events, as well as mutations of MTOR, NF1, NF2, MLH1, MLH3, MSH5, MSH6, ERBB2, EIF1AX, and USH2A (83). The era of precision medicine in cancer ushered a need for rapid genomic analysis of patient tumor samples because many mechanism-based treatments targeted specific oncogenic drivers of the disease. Although initially these assays were needed for implementation of clinical trials of these agents, many of these drugs have since become part of the standard of care for cancers of different lineages, including thyroid cancers.
To enable these treatments, cancer centers (84,85) and commercial entities (86) developed NGS panels covering the coding regions of a large number of genes known to be mutated across cancers of different types. Coupled to single institution small series of whole exome studies (87), the cumulative data arising from cancer exome profiling of large numbers of patient tumors have provided a deeper understanding of the genetics of advanced thyroid cancers (56,88). Aside from the role of MAPK pathway drivers across the entire spectrum of the disease, PDTC and ATC have a higher frequency of TERT promoter and TP53 mutations, as well as lesions of genes encoding for effectors in the PI3K/AKT/MTOR pathway and in chromatin remodeling complexes (Fig. 4). PDTC are also enriched for loss-of-function mutations of RBM10 (89), which encodes for an RNA binding protein that regulates alternative splicing of cassette exons (90).
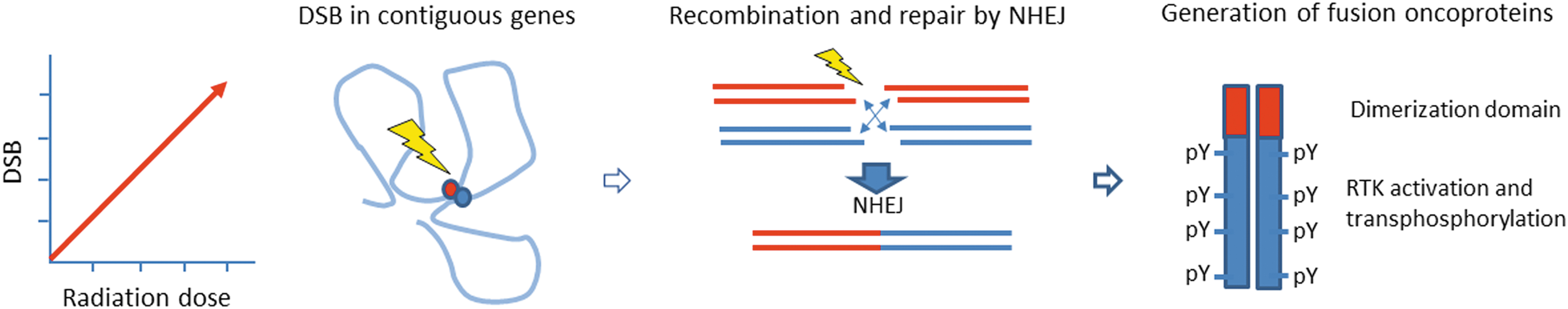
Mechanisms of radiation-induced thyroid cancer: Gene fusions are genetic hallmarks of thyroid cancer associated with exposure to ionizing radiation. Radiation exposure induces a dose-dependent increase in DNA double-strand breaks, short deletions, and simple/balanced structural variants, but not in single nucleotide variants. The radiation dose-dependent generation of fusion oncogenes is favored by spatial proximity of the participating genes and carries a signature of NHEJ at the fusion points. The most prevalent fusions involve RTK genes, which are commonly activated by recombination with a gene fragment encoding a protein dimerization domain. This drives homodimerization and activation by transphosphorylation of the cytoplasmic kinase domain of the RTK. NHEJ, nonhomologous end joining.
Whereas mutations of EIF1AX, a component of the translation preinitiation complex, are often seen in benign neoplasms and in about 30% of PTC as isolated events (19), in advanced thyroid cancers, they are almost invariably associated with RAS and TERT mutations and confer patients with decreased disease-specific survival (56,91). Interestingly, many of the mutations found in PDTC and ATC are present as subclonal events in PTC, supporting their role as determinants of tumor microevolution (19,56). In addition, PDTC and ATC are associated with copy number abnormalities, including among others chromosome 1q and 20q gain, and 13q loss, which associate with worse clinical outcomes (56). Loss of Chr 22q is one of few recurrent copy number abnormalities in PTC (19), and it is also highly prevalent in PDTC and ATC. PTCs with 22q LOH have a metastatic tropism to bone (92). Interestingly, three thyroid cancer tumor suppressor genes are encoded in 22q: CHEK2 (19), SMARCB1 (56,93), and NF2 (94,95).
Identification of Rare Thyroid Tumor Genes
In recent years, the landscape of genetic drivers of thyroid tumors has continued to fill in. Newly identified driver alterations activating the MAPK pathway include ROS1 fusions and MEK1 mutations in PTC (96,97). GLIS fusions, typically PAX8-GLIS3, have been found to be a genetic hallmark of a rare type of hyalinizing trabecular tumor (HTT), a rare thyroid tumor (98). They are characterized by a trabecular growth, prominent nuclear features characteristic of PTC and pronounced hyalinization, and had been suspected to be a variant of PTC. The identification of GLIS fusions in virtually all HTT, but not in PTC, provided the evidence that HTT is a distinct tumor unrelated to PTC.
Mutations in DICER1 gene coding for an endoribonuclease responsible for processing miRNAs were identified in thyroid cancers arising in children and adults, including those with sporadic tumors or tumors associated with an inherited DICER1 syndrome (99 –101). DICER1 somatic missense mutations cluster in metal-ion binding residues in the DICER1 RNAse IIIb domain, whereas germline mutations are typically nonsense mutations scattered along the gene. DICER1 mutations do not overlap with other common genetic alterations such as BRAF or RAS, supporting a distinct mechanism of transformation (100,101) Whereas the vast majority of DICER1-positive thyroid cancers are low-risk follicular variant PTC or FTC, rare cases of DICER1-driven PDTC occur in children and young adults, some of which develop in the setting of DICER1 syndrome (102). DICER1 joins the list of genes, which includes RET, PRKR1A, and PTEN, that when mutated in the germline predispose to thyroid neoplasia and that are also mutated in sporadic tumors (103).
Molecular Basis of Radiation-Associated Thyroid Cancer
Exposure to ionizing radiation is the only well-established risk factor for sporadic thyroid cancer. This includes exposure to medical external beam radiation as well as accidental exposures to radioiodine or γ-radiation after a nuclear weapon explosion or nuclear reactor accident (104). The link between radiation exposure and thyroid cancer, mostly PTC, has been known since 1950 after its demonstration in children exposed to therapeutic external beam radiation for benign conditions of the head and neck (105). It was not until the dramatic increase in incidence of thyroid cancer in children and young adults exposed to radiation after the accident at the Chernobyl nuclear power plant in April 1986 (106) that the genetic basis of radiation-induced thyroid cancer was uncovered.
Early candidate gene studies showed that PTC in children exposed to radiation after Chernobyl had a very high prevalence of RET fusions, mostly RET/PTC1 and RET/PTC3 (107 –110). Subsequent studies employing large cohorts of patients with post-Chernobyl PTC confirmed a high prevalence of RET and other fusions, such as NTRK1, in contrast to a low prevalence of BRAF point mutations in these cancers (111,112). The first fusion of BRAF in any cancer type (AKAP9-BRAF) was discovered in post-Chernobyl cancers, consistent with the association of these tumors with genomic rearrangements (113). Beyond post-Chernobyl cancers, RET fusions were also found to be prevalent in thyroid cancers from patients who received therapeutic external beam radiation (114).
RET/PTC1 and RET/PTC3 fusions could be induced experimentally by irradiating human thyroid tissue grafted into mice and in cultured human thyroid cells (115). Following DNA damage induced by ionizing radiation, RET fusions may be facilitated by close spatial proximity of chromosomal regions containing fusion partners in normal human thyroid cells in interphase (116). After the introduction of NGS-based genotyping, the association between gene fusions and radiation exposure was further supported by the high prevalence in Ukrainian children born before Chernobyl, in contrast with a higher frequency of point mutations in children born after the accident living in the same areas (117), as well as the demonstration of radiation dose dependence of gene fusions in children with post-Chernobyl cancer (118).
These findings have been confirmed and expanded in the recent large-scale genomic analysis of >400 post-Chernobyl thyroid cancers, demonstrating radiation dose-related increases in small clonal deletions and in fusion drivers that bear characteristics of nonhomologous end-joining repair of double-strand DNA breaks (119) (Fig. 5). Radiation dose dependence of these alterations was more pronounced for younger individuals at the time of exposure. The study also found that, similar to sporadic cancers, transcriptomic and epigenomic profiles were strongly associated with driver alterations, although they did not correlate with radiation dose (119). A related study of children born after the Chernobyl accident to parents exposed to radiation reported no significant increase in germline de novo mutations, suggesting minimal effect of the parents' radiation exposure on health of subsequent generations (120).
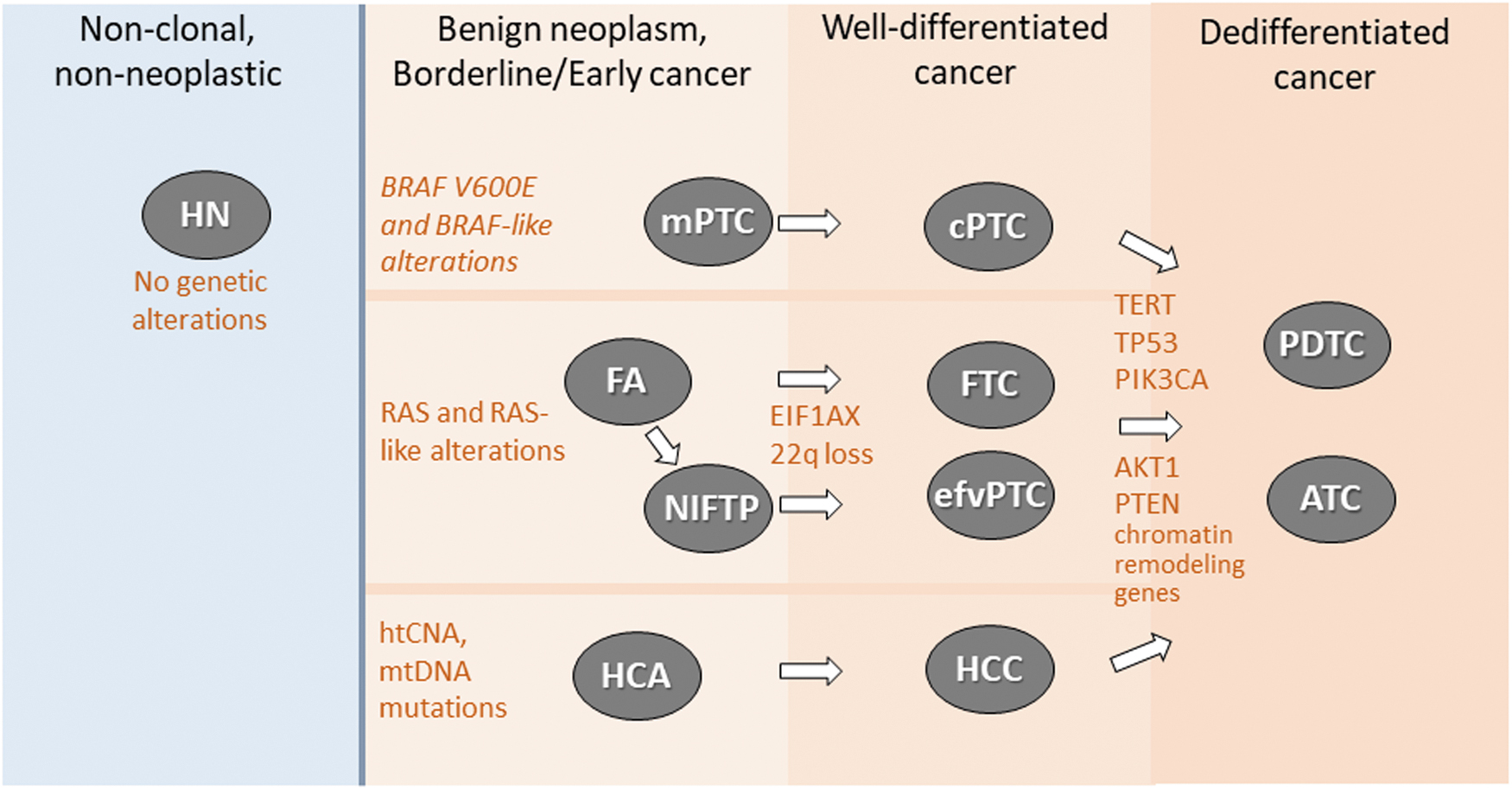
Molecular classification of thyroid nodules. Most thyroid nodules are HN that carry no clonal genetic alterations. Follicular cell-derived thyroid tumors develop via three distinct molecular pathways initiated by BRAFV600E-like alterations, RAS-like alterations, or mtDNA mutations and chromosomal copy number abnormalities leading to genome haploidiation-type copy number alterations (htCNA). RAS-like encapsulated follicular-patterned tumors, including FTC, invasive efvPTC, and htCNA/mtRNA-driven encapsulated HCC likely develop from benign/preinvasive precursors: that is, FA/NIFTP and HCA, respectively. BRAF-like cPTC lack a benign precursor and develop by growth of a micropapillary thyroid cancer (mPTC). Progression of RAS-like tumors to well-differentiated cancer frequently involves EIF1AX mutations and/or 22q loss, whereas conversion of cancers initiated by all three molecular pathways to PDTC and ATC involve the accumulation of additional mutations in TERT, TP53, PI3K pathway mutations, and alterations of chromatin remodeling genes. ATC, anaplastic thyroid cancer; cPTC, classic papillary thyroid carcinomas; efvPTC, encapsulated follicular variant papillary thyroid carcinoma; FA, follicular adenoma; FTC, follicular thyroid carcinoma; HCA, Hurthle cell adenoma; HCC, Hurthle cell (oncocytic) cancer; HN, hyperplastic nodules; htCNA, genome haploidiation-type copy number alterations; mPTC, micropapillary thyroid cancer; mtRNA, mitochondrial RNA; NIFTP, noninvasive follicular tumor with papillary-like nuclear features; PDTC, poorly differentiated thyroid carcinoma; PI3K, phosphatidylinositol 3-kinase.
Genetic Predisposition to Thyroid Cancer
In addition to familial MTC in patients with MEN syndromes, several familial diseases increase the risk of nonmedullary thyroid cancer. The association between familial adenomatous polyposis/Gardner syndrome and thyroid cancer was established in the 1960s, and in 1991, this syndrome was linked to a germline mutation of the APC gene (121). Thyroid cancers developing in these patients have distinct microscopic features initially described as cribriform-morular variant PTC (122), recently renamed as cribriform-morular thyroid carcinoma.
Thyroid nodularity and cancer are a common manifestation of the Cowden syndrome, and several other rare syndromes collectively known as the PTEN hamartoma tumor syndrome, which is caused by germline mutations of the PTEN tumor suppressor gene (60,123). An increased risk of thyroid cancer has also been found in patients affected by the Carney complex caused by inactivating mutations in the PRKAR1A gene (124,125), and by Werner syndrome linked to the WRN gene (126). More recently, thyroid nodules and cancer have been found to be a manifestation of DICER1 syndrome caused by inactivating mutations in the DICER1 gene, which is essential for maturation of miRNAs (127,128).
Translation of Genomics into Clinical Practice: Molecular Diagnostics of Thyroid Nodules
The information on genomic alterations in thyroid cancer gained over the past four decades has been translated into clinical practice for patients with thyroid nodules and cancer. Fine-needle aspiration (FNA) cytology of sonographically suspicious nodules provides a reliable diagnosis of benign or malignant nodule in ∼80% of cases (129). In the remaining 20%, the cytology is indeterminate (130). In the pre-molecular era, most nodules with indeterminate cytology would prompt diagnostic lobectomy, which yielded cancer in only about one third of cases. The ability to detect gene mutations in thyroid FNA samples was demonstrated in the early 1990s (131). However, it took more than a decade for the progress in technology coupled to a more comprehensive understanding of the genetic drivers in thyroid cancers to enable the development of the first-generation genetic tests for diagnosing PTC and FTC in FNA samples.
Initial experience with panels of six to seven genes (BRAF, NRAS, HRAS, KRAS, RET/PTC1, RET/PTC3, PAX8-PPARG) analyzed by single-gene PCR-based assays was reported in 2009 and 2010 (132,133). These relatively limited panels had a high specificity but lacked the sensitivity required to avoid diagnostic surgery. In the ensuing years, second-generation molecular diagnostic tests have been introduced into clinical practice, which include larger panels composed of different combinations of DNA, RNA, and miRNA markers (134 –138). These tests, offered commercially and broadly available in the United States, had either high sensitivity or high specificity but did not achieve both.
Finally, over the last 5 years, most comprehensive DNA/RNA or RNA/NGS-based tests have been extensively validated to offer high sensitivity and fairly high specificity for detecting follicular cell-derived thyroid cancers as well as MTC and parathyroid nodules that masquerade cytologically as follicular cell-derived nodules (139,140). The introduction of molecular tests has resulted in a significant decrease in the rate of diagnostic surgeries for patients with indeterminate cytology thyroid nodules (141).
Molecular Diagnostics as a Guide for Systemic Therapy of Advanced Metastatic Thyroid Cancers
The first generation of effective systemic therapies for RAI-refractory metastatic differentiated thyroid cancer and MTC is small-molecule multikinase inhibitors such as lenvatinib, sorafenib, and pazopanib that act primarily by inhibiting VEGF receptor activation and inhibiting angiogenesis, regardless of the nature of the oncogenic driver of the disease (142,143). Similarly, the multikinase inhibitors vandetanib and cabozantinib, which do target RET kinase activity, also benefit RET-wild-type MTC patients because of their inhibition of VEGFR (144,145). Hence, when these drugs were the only available treatment option for patients, determination of tumor genotype did not meaningfully contribute to disease management.
The landscape of thyroid cancer therapies has evolved dramatically in the past 5 years, such that tumor genotype is now critical to determine the best treatment option for patients with advanced locally recurrent or metastatic disease (146). The recent approval of the highly selective RET kinase inhibitors selpercatinib and pralsetinib requires tumor genotyping, as they are indicated solely for MTC with RET mutations or cancers driven by RET fusions. The rationale for tumor genotyping is further buttressed by the fact that thyroid cancers harboring other receptor tyrosine kinase (RTK) fusions, such as of NTRK3, NTRK1 or ALK, are candidates for treatment with selective inhibitors of these mutant oncoproteins. The first line of systemic treatment for patients with BRAFV600E-mutant ATC is the combination of the RAF kinase inhibitor dabrafenib with the MEK inhibitor trametinib, which has also shown efficacy in the neoadjuvant setting. The recent FDA approval for combined RAF and MEK inhibition now extends to any BRAFV600E -mutant cancers, regardless of their origin.
Summary and Future Directions
Differentiated thyroid cancers, MTC, and Hurthle cell (oncocytic) cancer (HCC) are arguably among the best characterized human cancer types with respect to the genetic drivers responsible for their development. This has already had a transformational impact on diagnostics of thyroid nodules and on treatment for the subset of these patients whose tumors recur, become refractory to radioiodine, or develop distant metastatic disease. We have also gained information on the more complex genomic and transcriptomic features of PDTC and ATC. Although the progress has been remarkable, there is still much to learn.
Our understanding of advanced disease is based on cancer exome panels rather than whole exome or whole-genome sequencing, and the studies available so far have not integrated the multidimensional molecular data required to provide a deeper understanding of their biology. There is also scant information on the microevolution of metastatic thyroid cancer. The use of plasma free DNA assays to monitor response to therapy and acquisition of resistance in patients with metastatic thyroid cancer on systemic therapies is still in its infancy. Moreover, the clonal, transcriptional, and compositional heterogeneity of advanced disease poses challenges that will require unraveling mechanisms and pathways that may not necessarily be dependent directly on the genetic drivers of the disease.
Footnotes
Authors' Contributions
J.A.F. and Y.E.N. co-wrote and contributed equally to this review.
Author Disclosure Statement
Y.E.N. owns IP and receive royalties related to ThyroSeq from the University of Pittsburgh and serves as a consultant for Sonic Healthcare USA. J.A.F. reports no disclosures.
Funding Information
This study was supported by the National Institutes of Health (NIH) R01 grants CA50706-27, CA255211-02, and CA249663-02 (to J.A.F.) and NIH CA181150 (to Y.E.N.).